Bioquímica/Imprimir
Bioquímica

O que é a Bioquímica?
editar


Todos os sistemas vivos, do mais simples ao mais complexo, dependem de reações químicas fundamentais à sua sobrevivência. Os organismos crescem e reproduzem-se através da multiplicação celular, mas de onde provêm os constituintes das células? Como os nutrientes absorvidos por um organismo são transformados em tecidos tão diversificados, como músculos ou o sangue? E como podem organismos tão pequenos como uma única célula ter uma vida independente?
As respostas a estas questões estão na base do desenvolvimento da Bioquímica. A Bioquímica pode ser definida como o estudo da química de organismos vivos e da química relacionada com estes. Forma uma ponte entre a Biologia e a Química pois estuda como complexas reações e estruturas químicas originam vida e processos relacionados com a vida. É considerada por vezes um ramo híbrido da Química Orgânica, especializado nos processos químicos que ocorrem nos organismos vivos, mas na realidade a Bioquímica não pode ser considerada um ramo de apenas a Biologia ou da Química - a Bioquímica incorpora todas as interações existentes da menor molécula biológica à maior célula existente.
De uma forma essencial, a Bioquímica estuda a estrutura e função de componentes celulares (como enzimas e organelas) e os processos efetuados por moléculas de diferentes dimensões, como as proteínas, ácidos nucleicos, glicídios e lípidos, entre outros. Segundo a teoria da evolução, os organismos existentes na atualidade descendem de um antepassado comum, explicando a semelhança dos processos bioquímicos existentes em todos os organismos vivos.
Posto de forma simples, a Bioquímica é a Química da Vida.
Métodos de estudo
editarUm dos métodos de estudo mais empregues em Bioquímica é a aproximação reducionista a um problema. É usual fazer-se a purificação de componentes dos sistemas vivos, como proteínas, para estudar as suas propriedades de forma isolada. Esta aproximação é muito útil para o conhecimento profundo de aspectos estruturais e funcionais dos componentes dos sistemas vivos, mas tem a desvantagem de impedir o estudo de interações que ocorram in vivo: numa célula, nenhum componente se encontra isolado. Por isso, também existem métodos de estudo holísticos, que tentam determinar as propriedades de um sistema como um todo; um exemplo é o estudo do comportamento de vias metabólicas inteiras, em vez de estudar cada enzima que delas fazem parte.
A Bioquímica é uma ciência essencialmente experimental, mas com o desenvolvimento de mais e melhores ferramentas computacionais e matemáticas, existe também uma grande área de investigação bioinformática associada. Algumas das aplicações mais populares incluem a previsão de interação entre proteínas, a modelação da sua estrutura tridimensional, a comparação de sequências proteicas e nucleotídicas e a aplicação de modelos estatísticos a amostras reais.
História
editarA Bioquímica tem as suas raízes na história da Química, em particular no interesse do homem em saber que transformações ocorriam nos organismos vivos, responsáveis pela sua origem, crescimento e metamorfose. As questões colocadas por aqueles que procuraram compostos na Natureza que curassem doenças, que se interrogaram sobre a fisiologia do corpo humano, que usaram processos naturais como a fermentação de cervejas e que observaram a decomposição da matéria orgânica, entre outros, lançaram as bases da Bioquímica tal como é conhecida na atualidade.
Na Antiguidade
editar
A religião taoísta desenvolvida há mais de 2000 anos na China concebia o mundo como dividido em dois opostos, Yin e Yang, que, na luta para se manterem em equilíbrio, gerariam os cinco elementos água, terra, fogo, madeira e metal, que constituiriam todas as coisas. Os chineses preparavam então elixires contendo compostos de modo a equilibrar Yin e Yang no corpo; esta busca de elixires levou à descoberta de medicamentos e processos de fermentação. Experiências com fluidos corporais levaram provavelmente à descoberta de hormônios sexuais. Este tipo de alquimia refletia então as práticas médicas/farmacêuticas da época.
Existem também registros que as antigas civilizações egípcias e babilônicas praticavam a extração de substâncias com propriedades farmacológicas e de perfumes a partir de plantas.
Na Grécia Antiga, Empédocles postulou no século V a.C. o mundo como comporto por elementos, de forma similar às ideias taoístas. Desta feita, seriam quatro elementos: fogo, ar, água e terra. A escola aristotélica desenvolveu esta ideia, que foi no entanto rebatida por Demócrito. Demócrito introduziu o conceito de atomicismo, de que a diferença entre pequeníssimas partículas indivisíveis, que juntas constituiriam toda a matéria, explicariam as diferenças macroscópicas desta. Esta teoria atômica da constituição da matéria, comparável com a teoria moderna, foi no entanto esquecida até ao século XVI.
É também sabido que os povos árabes possuem uma longa tradição de conhecimentos farmacológicos, mas foram os antigos Gregos quem desenvolveram a alquimia, uma das bases da Química moderna, e portanto também da Bioquímica, com a preparação de poções e tinturas a partir de minerais e plantas.
Idade Moderna
editar
Paracelso, no século XVI, formulou o universo como sendo regulado por leis químicas, em que os processos químicos serviriam de base às transformações observadas na Natureza. Também estabeleceu a ideia de que as doenças deveriam ser tratadas usando compostos químicos e que a natureza das próprias doenças estaria ligada à putrefação dos "dejetos" provenientes de processos químicos. A utilização da Química para fins medicinais foi denominada "iatroquímica" por Paracelso.
Apesar de Paracelso misturar diversos conceitos místicos e alquimistas nos seus ensinamentos, a utilização de compostos químicos com fins farmacêuticos generalizou-se, especialmente no século XVII, devendo-se parte desta generalização da iatroquímica a w:Jan Baptista van Helmont, discípulo de Paracelso. Numa publicação póstuma (1648), van Helmont descreve uma experiência em que apenas água havia sido adicionada a um jovem salgueiro plantado em terra previamente seca num forno. O salgueiro cresceu sem haver apreciável variação da massa da terra e van Helmont atribuiu este crescimento à transformação de água em madeira, casca e raízes. Embora van Helmont tenha retirado as conclusões erradas das suas experiências, houve de sua parte uma tentativa de planear meticulosamente e de quantificar as transformações observadas. Outras observações de van Helmont relevantes à história da Bioquímica incluem a sua descrição da digestão como um processo fermentativo usando ácido e a excreção de líquidos alcalinos no corpo humano, nomeadamente a bílis.
As primeiras experiências sobre o metabolismo animal conduzidas de forma controlada foram publicadas por Santorio Santorio em 1614 no seu livro Ars de statica medecina, no qual Santorio descreveu como determinou o seu próprio peso antes e depois de comer, beber, dormir, trabalhar, ter relações sexuais, jejuar e excretar. Ele descobriu que a maior parte da comida ingerida era perdida no que ele denominou de "perspiração insensível".
Franciscus Sylvius, discípulo de van Helmont, ampliou o conceito de digestão, englobando a participação da saliva e sucos pancreáticos e caracterizando o processo digestivo como uma neutralização entre ácidos e bases. Sylvius considerou que também noutros processos fisiológicos ocorreria neutralização e que, deste modo, as doenças surgiriam de situações em que existisse um excesso de ácido ou de base no corpo.
A influência do atomicismo
editar
A ideia de que toda a matéria seria composta por unidades de tamanho diminuto, sendo por isso invisíveis se pensadas isoladamente, ganhou força no século XVII. A invenção do microscópio composto, por Robert Hooke, permitiu as primeiras observações de células. Robert Boyle considerou a matéria como composta por corpúsculos; Descartes apoia a teoria atomicista. Boyle afirma que as propriedades da matéria são explicáveis pelas propriedades físicas (tamanho e forma) dos corpúsculos, assim como pelo seu movimento, e que as transformações químicas são produzidas pela interação mútua entre essas diminutas entidades.
O conceito de oxidação
editarAté aos trabalhos de Antoine-Laurent Lavoisier, persistiram ideias como a dos elementos aristotélicos (ou variações desta; uma ideia particularmente persistente foi a do fogo como elemento de transformação) e a teoria do flogisto. Lavoisier explicou pela primeira vez a oxidação de metais e não-metais pelo oxigênio e alargou o conceito de oxidação aos processos fisiológicos: o oxigênio respirado seria utilizado na "combustão" do carbono contido em alimentos, formando-se dióxido de carbono, expulso na expiração, e calor, que explicaria o calor interno dos animais. Uma parte do ar que não seria respirável, o nitrogênio, seria expulso sem alteração.
Nos finais do século XVIII, Joseph Priestley, que dedicou uma grande parte da sua vida ao estudo dos gases,descobriu que as plantas produziam "ar desflogistificado", isto é, contendo oxigênio, na presença de luz; foi na mesma época que Jan Ingenhousz e Jean Senebier formularam uma teoria para a fotossíntese.
Idade Contemporânea
editarA influência da Química-Física
editarQuando, nos finais do século XIX, começou a haver um interesse na química de soluções, e a Química-Física ascendeu à categoria de ramo independente da Química, surgiu também um renovado interesse pelo estudo da concentração do íon hidrogênio (H+) e a sua relação com o pH. O dinamarquês Søren Sørensen sugeriu em 1909 utilizar o negativo do logaritmo da concentração de H+, -log[H+], originando a escala de pH tal como é conhecida na atualidade, uma ferramenta de auxílio na determinação da força de ácidos e bases. O conceito de pH foi rapidamente assimilado nos estudos bioquímicos, pois eram então conhecidas as capacidades de tampão de sistemas fisiológicos, evitando extremos de acidez ou alcalinidade.
O fim do vitalismo
editarNo século XIX, aquando do estudo da fermentação de açúcar a álcool em leveduras, Louis Pasteur concluiu que a fermentação era catalisada por uma força vital dentro das células, a que chamou "fermentos". Pensava-se então que os fermentos funcionavam apenas dentro de organismos vivos.

Originalmente acreditava-se que a vida não era assunto para a ciência. Acreditava-se que apenas os seres vivos podiam criar as "moléculas das vida" (a partir de moléculas já existentes). Este pensamento começou a mudar a partir do ano de 1828 quando Friedrich Wöhler publicou um trabalho sobre a síntese de ureia, provando que compostos orgânicos podiam ser criados artificialmente e derrubando o vitalismo como base teórica para a distinção entre a matéria animada e inanimada.
A Bioquímica moderna
editarTalvez o fator crucial para o surgimento da Bioquímica tenha sido a descoberta da primeira enzima em 1833, que na época recebeu o nome "diastase" (hoje chamada "amilase"); quem a descreveu foi Anselme Payen. Em 1896, Eduard Buchner contribuiu para a Bioquímica descrevendo pela primeira vez um complexo processo bioquímico fora da célula - a fermentação alcoólica de extratos celulares de fermento - o que lhe valeu o Prêmio Nobel da Química de 1907.
Outro avanço importante na Bioquímica foi a demonstração da natureza proteica das enzimas por James B. Sumner, um assunto anteriormente controverso, ao conseguir cristalizar primeiro a enzima urease e, mais tarde, a catalase. A cristalização de proteínas permitiu a aplicação de técnicas de raios-X para a determinação de estruturas tridimensionais proteicas, algo conseguido pela primeira vez com a enzima lisozima. Mais tarde, e após os estudos de Linus Pauling sobre a natureza da ligação química e a estrutura das hélices alfa proteicas, James Watson, Francis Crick e Maurice Wilkins publicaram a estrutura tridimensional da dupla hélice do DNA.
Embora o termo "bioquímica" tenha sido usado pela primeira vez em 1882, é mais aceito que a criação formal deste termo tenha ocorrido em 1903 pelo químico alemão Carl Neuberg. Anteriormente essa área de ciência era denominada "química fisiológica". Desde a metade do século XX em diante, a bioquímica avançou muito; esse avanço foi possível graças ao desenvolvimento de novas técnicas como a cromatografia, difração de raio X, espectroscopia de ressonância magnética nuclear (RMN), microscopia eletrônica e simulação da dinâmica molecular. Estas técnicas permitiram a descoberta e análise detalhada de moléculas e vias metabólicas celulares, que possibilitaram, por exemplo, elucidar a glicólise ou o ciclo de Krebs (ciclo do ácido cítrico).
Hoje, os resultados e princípios bioquímicos são empregados em muitas outras áreas que vão da genética à biologia molecular, da agricultura à medicina.

O conceito de que a célula e a unidade básica da vida já está bem impregnado em nossas mentes, de igual forma também sabemos que, pelo menos até o presente momento, a vida nada mais é do que um complexo e não totalmente elucidado conjunto de reações químicas. Embora estejamos muito longe de desvendar a "vida" em seu atual estagio, quem nunca se perguntou - "Como ela surgiu?". Muitos cientista já fizeram a mesma pergunta, sendo que alguns até tentaram criar teorias que explicassem tal acontecimento. Na década de 30, dois cientistas (Alexandre Oparin e J. B. S. Haldane) de forma independente elaboraram uma teoria em comum, com base nos prováveis constituintes da atmosfera primitiva (H2O, N2, CO2, CH4, NH3, SO2 e possivelmente H2) eles chegaram a conclusão de que a radiação ultravioleta e/ou descargas elétricas faria com que as moléculas simples presente na atmosfera reagissem entre si dando origem a moléculas mais complexas, inclusive moléculas orgânicas como, por exemplo, aminoácidos. Tal teoria pode ser comprovada por um experimento que reproduziu as hipotéticas condições da atmosfera primitiva, este experimento foi realizado em 1953 por Staley Miller e Harold Urey. A partir deste acontecimento inúmeras outras teorias foram elaboradas para explicar como as células puderam se originar de tal situação, no entanto tais mecanismos são ainda muito obscuros, e devido a esse motivo não vamos nos ater a este detalhes. No entanto por qualquer que seja os caminhos seguidos pela evolução da vida está muito claro que há uma similaridade entre os seres vivos atuais, e graças a essa similaridade é possível, através de formas de vida mais simples, desvendar alguns dos "segredos da vida" que nos permite desenvolver estratégias para vivermos mais e melhor.
Organização da Vida
editarOs domínios da Vida
editar
Os organismos vivos apresentam uma grande diversidade morfológica e funcional. Podem ser constituídos por apenas uma célula ou terem milhões de células organizadas em tecidos diferenciados; podem criar o seu próprio alimento ou necessitar de o obter de alguma fonte; podem depender do oxigênio ou nem sequer tolerar a sua presença. De forma a sistematizar os organismos do nosso planeta, é feita a divisão dos mesmos de acordo com estas e outras características comuns. O ramo científico que estuda a classificação dos seres vivos é a taxonomia.
Na atualidade, é geralmente aceita a divisão de todos os organismos em três domínios, proposta por Carl Woese:
- o domínio Eukarya inclui todos os organismos eucariontes;
- o domínio Eubacteria ou simplesmente Bacteria inclui todas as bactérias (procariontes);
- o domínio Archaea (anteriormente designado "Archaebacteria") inclui procariontes com características filogenéticas (relações evolucionárias obtidas por análise genómica) diferentes da bactérias.
Nesta classificação não se incluem os vírus. É ainda assunto de debate se os vírus devem ser classificados como sistemas vivos ou não; apesar de conterem material genético, poderem reproduzir-se e apresentarem um "ciclo de vida", não são capazes de o suster de forma independente e não têm a característica organização interna dos organismos vivos. Também os priões, partículas proteicas com características virais, estão fora desta classificação proposta por Woese.
Tanto as bactérias como as arqueas são por vezes englobadas num super-reino Prokaryota (procariontes; por vezes designado Monera, um termo obsoleto) quando se considera uma divisão em duas categorias (sendo a outra a Eukaryota). No entanto, os estudos filogenéticos de Woese e posteriores demonstram que as arqueas e as bactérias têm ramos de evolução distintos; em adição, existem algumas características morfológicas e metabólicas mais próximas entre arqueas e eucariontes que entre arqueas e bactérias. É também atualmente referido o super-reino Acytota (organismos acelulares), englobando os vírus e priões.
Um terceiro sistema de classificação geral dos seres vivos consiste em considerar a existência de seis reinos:
- Archaebacteria - correspondente ao domínio Archaea.
- Eubacteria - as verdadeiras bactérias.
- Fungi - fungos.
- Plantae - plantas.
- Animalia - animais.
- Protista - eucariontes unicelulares e algas sem verdadeiros tecidos.
No sistema dos três domínios de Woese, Fungi, Plantae e Animalia são reinos pertencentes ao domínio Eukarya; o reino Protista engloba diversos grupos filogeneticamente diversos mas que não se encaixam nos restantes reinos eucarióticos.
A classificação hierárquica biológica
editar
A divisão dos organismos em domínios é muito geral e existe necessidade de os organizar de forma mais sistemática. O primeiro sistema taxonômico foi desenvolvido pelo cientista sueco Lineu, que estabeleceu a seguinte hierarquia:
Assim, um reino poderia conter diversos filos; cada filo, diversas classes, etc., até se chegar à classificação por espécies, que distingue organismos atribuindo-lhe um nome binomial em latim, composto pelo nome do gênero correspondente e um epíteto específico. Lineu usou características morfológicas para distinguir espécies; muita da classificação por ele feita subsiste até hoje, classificação esta baseada não só em características morfológicas mas também (e especialmente) em características filogenéticas. O sistema de classificação sofreu alguns refinamentos (como por exemplo a existência de supergrupos ou subgrupos, como sub-ordens ou superfamílias).
Bases para a classificação
editarA divisão entre organismos eucarióticos e organismos procarióticos deve-se às diferenças fundamentais das células que constituem estes dois grupos. As células eucarióticas possuem um núcleo bem definido contendo material genético, delimitado por uma membrana; possuem diversos organelas também delimitados por membranas, nos quais ocorrem processos metabólicos especializados - existe portanto compartimentalização na célula. Tal nível de compartimentalização não existe em células procarióticas. as diferenças entre os diferentes tipos de células são exploradas no capítulo sobre organização celular.
Após os trabalhos de Woese, que analisou o RNA ribossomal de diversos procariontes, tornou-se óbvia uma distinção entre diferentes membros deste grupo, surgindo a necessidade de o separar em dois domínios, Archaea e Eubacteria. As arqueas possuem algumas características bem distintas das verdadeiras bactérias, incluindo vias metabólicas modificadas ou únicas e a presença de lípidos membranares distintos dos bacterianos. Mais curiosamente, possuem algumas características mais próximas das dos eucariontes que de procariontes, como o mecanismo de transcrição e tradução do DNA. As arqueas são organismos extremófilos, ou seja, vivem em condições consideradas extremas para outros organismos, como alta salinidade, temperaturas muito altas (como as presentes em hotspots) ou muito baixas (encontra-se arqueas no gelo antártico) e ambientes de extrema acidez. Embora a classificação filogenética dos três domínios seja ainda algo controversa, é aceite que existiu primeiro uma divergência que criou o grupo das bactérias e mais tarde uma segunda divergência do grupo das não-bactérias que originou as arqueas modernas e os eucariontes. Também se pensa que as arqueas terão sofrido poucas modificações ao longo da evolução, por se encontrarem preferencialmente em nichos ecológicos com condições semelhantes aos dos primórdios da vida na Terra.
Quase todos os eucariontes conhecidos dependem da presença de oxigênio para o seu metabolismo normal, ou seja, são organismos aeróbios. O oxigênio é necessário na fosforilação oxidativa como aceitador final de elétrons. Em contraste, os procariontes apresentam uma grande diversidade de metabolismo energético, podendo classificar-se de diferentes formas quanto ao aceitador final de elétrons e quanto à fonte de carbono usada para o seu desenvolvimento:
- Organismos fototróficos usam a energia luminosa como fonte energética. Dentro destes, existem os:
- Organismos fotoautotróficos, que usam o dióxido de carbono (CO2) como fonte primária de carbono; nestes incluem-se as cianobactérias mas também, por definição, as plantas.
- Organismos foto-heterotróficos, que usam compostos orgânicos como fonte primária de carbono. Incluem alguns tipos de bactérias (como as bactérias púrpura).
- Organismos quimiotróficos usam a energia retirada do catabolismo de compostos químicos. Dentro destes, existem os:
- Organismos quimio-heterotróficos: usam compostos orgânicos como fonte de carbono) e dividem-se por sua vez (dependendo da fonte de energia) em:
- Organismos quimiolitotróficos, que usam compostos inorgânicos como fonte de energia. Exemplos incluem as bactérias redutoras de sulfato, que usam este anião.
- Organismos quimiorganotróficos, que usam compostos orgânicos como fonte de energia. Nestes incluem-se a maioria das bactérias, assim como todos os eucariontes não fototróficos.
- Organismos quimioautotróficos: utilizam como fonte energética compostos inorgânicos.
- Organismos quimio-heterotróficos: usam compostos orgânicos como fonte de carbono) e dividem-se por sua vez (dependendo da fonte de energia) em:
Os organismos que não usam o oxigênio molecular como aceitador final de elétrons (ou seja, que não respiram oxigênio mas sim outro composto químico) são denominados anaeróbios. Os anaeróbios que não suportam sequer a presença de oxigênio são anaeróbios obrigatórios. Alguns procariontes apresentam características mistas: podem por exemplo usar oxigênio nalgumas ocasiões mas também utilizar metabolismo anaeróbio quando a quantidade de oxigênio é limitante (aeróbios facultativos); ou então necessitar de gás carbõnico para o seu metabolismo mas a uma pressão parcial menor que a atmosférica (microaerofílicos). Existem também designações mais específicas para um determinado tipo de metabolismo; por exemplo, organismos metanogênicos são aqueles que sintetizam metano como produto da respiração; bactérias sulfurosas usam compostos de enxofre como fonte de energia metabólica, etc.
Como já anteriormente abordado neste livro, não é possível explicar com grande clareza como as células surgiram, mas se observarmos a natureza perceberemos, sem qualquer dúvida, que tudo que é ou que julgarmos ser vivo é composto por células. Portanto para entendermos um pouco mais sobre a vida, nada melhor do que estudarmos as células.
De acordo com características morfológicas podemos dividir as "células" em dois grandes grupos: procariontes e eucariontes. Devido ao nível de complexidade vamos começar abordando as células procariontes e depois as eucariontes.
Células procariontes
editar
O termo procarionto é derivado das palavras gregas pro que significa anterior, antes, e karyon que significa noz ou amêndoa - núcleo (anterior ao núcleo). São organismos unicelulares que não possuem um envoltório nuclear, cujo material genético encontra-se disperso no citoplasma como o próprio termo sugere. Esta definição engloba todos os organismos dos domínios Bacteria e Archaea. Para descrevermos a estrutura de uma célula procarionte nada melhor do que começarmos por seu envoltório celular.
Envoltório celular
editarO envoltório celular bacteriano é constituído por uma membrana interna (membrana plasmática - semelhante a dos eucariotos) e por uma segunda camada (a parede celular propriamente dita) que é composta principalmente por peptidoglicano. No caso das bactérias gram-negativas ainda há uma terceira camada (membrana externa) que é semelhante à membrana interna, no entanto muito mais permeável. Das estruturas mencionadas anteriormente apenas a membrana plasmática não faz parte da parede celular bacteriana.
Parede celular
editarPodemos considerar a parede celular sendo uma, se não a mais, importante estrutura para as bactérias, sendo esta o alvo de muitos antibióticos que, por exemplo, inibem sua formação. Por ser muito resistente, permitindo que a bactéria sobreviva em ambientes muito hostis, esta exerce uma força contrária à da osmose evitando que a bactéria estoure; a parede celular de algumas bactérias resiste a uma pressão de até 20 atm. Além do mais, é responsável pela forma (morfologia) bacteriana de uma maneira análoga a um pneu, e é característico de cada espécie bacteriana que pode ser semelhantes entre algumas espécies ou em alguns casos muito diferentes, permitindo assim uma forma de classificação bacteriana (para melhor esclarecimento clique aqui). Deve-se levar em consideração que alguns procariontes não possuem parede celular, como os micoplasmas.
Saiba mais
editarHá mais de cem anos, Hans Christian Gram (médico dinamarquês) desenvolveu uma técnica de coloração que hoje é nomeada Técnica de Gram. Não vamos nos ater aos detalhes da técnica, mas esta permitiu dividir as bactérias em dois grandes grupos: as bactérias gram-positivas e gram-negativas. Por este método não é possível caracterizar a estrutura bacteriana responsável pela coloração. Com o passar dos anos foram desenvolvidas novas tecnologias que permitiram identificar a ultra-estrutura bacteriana (microscopia eletrônica e desenvolvimento de novas técnicas de análise bioquímicas). Hoje sabemos que isto se deve a diferenças na ultra-estrutura da parede celular bacteriana.
Membrana plasmática
editarÉ uma dupla camada de fosfolipídios e proteínas, esta última tem várias funções como permeabilidade seletiva, produção de energia, etc. Delimita o que está dentro ou o que está fora da célula (para mais detalhes clique aqui). Quando analisada por intermédio da microscopia eletrônica, é possível visualizar invaginações desta membrana. A estrutura recebe o nome de mesossoma e, embora seja lhe sejam atribuídas funções na respiração e divisão celular, alguns autores afirmam que esta não possui função alguma e que seja apenas um simples artefato de preparação para visualização na microscopia eletrônica.
Hialoplasma ou citoplasma
editarÉ um líquido com consistência de gel, contendo sais, glicose e outros açúcares, proteínas funcionais e várias outras moléculas orgânicas. Contém também ARNm (ARN mensageiro) e ribossomas. Os ribossomas procariotas são bastante diferentes dos eucariotas (essas diferenças foram usadas para desenvolver antibióticos usados para afetar exclusivamente os ribossomas das bactérias). No citoplasma também está presente o seu único cromossomo; os procariontes podem possuir material genético extracromossomal, denominado plasmídeo, que são pequenas moléculas de DNA circular que normalmente contêm genes que conferem resistência a antibióticos. Os procariontes podem ter mais de uma cópia de plasmídeo.
Células eucariontes
editar
A palavra Eucarionte é derivada do grego eu, que significa verdadeiro e karyon, que significa noz ou amêndoa - núcleo. Como o próprio nome sugere, inclui todos os seres vivos com células que possuem núcleo que delimita o seu material genético do citoplasma. Além disso as células eucariontes possuem várias organelas.
Quando comparado com as células procariontes, os eucariontes são muito mais complexos, possuem várias organelas e a maioria das reações ocorre em compartimentos próprios. A transdução de sinal é muito mais sofisticada e o seu material genético está numa forma mais compactada do que em procariontes.
As células eucarióticas apresentam várias diferenças entre si. Se analisarmos uma célula animal, uma vegetal e um fungo, encontraremos diferenças significativas, no entanto todas essas apresentam características em comum; começaremos com as estruturas comuns a essas células e posteriormente abordaremos suas diferenças.
Membrana plasmática
editarA membrana celular é a estrutura que estabelece a fronteira entre o meio intracelular e o meio extracelular; também controla a entrada e saída de substâncias de uma forma muito seletiva. Sua estrutura, como mostra a figura ao lado, é composta por uma dupla camada lipídica sendo que nesta estão envolvidas proteínas, que têm inúmeras funções que vão de transporte a adesão celular.
Citoplasma
editar
É o espaço intracelular entre a membrana plasmática e o envoltório nuclear. O citoplasma é preenchido por uma matéria coloidal e semi-fluída onde estão suspensos as organelas celulares. O citoplasma não inclui o núcleo celular, cujo interior é formado por nucleoplasma.
O componente não solúvel do citoplasma é constituído por organelas: mitocôndrias, cloroplastos, lisossomas, peroxissomas, ribossomas, vacúolos, citoesqueleto e outras estruturas membranares (complexo de Golgi e retículo endoplasmático).
Núcleo
editar
É uma estrutura presente nas células eucariontes, que contém o DNA da célula. Foi descoberto em 1833 pelo pesquisador escocês Robert Brown. É delimitado pelo envoltório nuclear, e se comunica com o citoplasma através dos poros nucleares. O núcleo possui duas funções básicas: regular as reações químicas que ocorrem dentro da célula, e armazenar as informações genéticas da célula. O seu diâmetro pode variar de 11 a 22,25 μm. Dentro do núcleo ainda podemos encontrar uma estrutura denominada nucléolo, que é responsável pela produção de subunidades dos ribossomos. Sua posição é geralmente central, acompanhando o formato da célula, mas isso pode variar de uma para outra. Nos eritrócitos dos mamíferos, o núcleo está ausente.
O envoltório nuclear é responsável tanto por separar as reações químicas que ocorrem dentro do citoplasma daquelas que ocorrem dentro do núcleo, quanto por permitir a comunicação entre esses dois ambientes. Essa comunicação é realizada pelos poros nucleares que se formam da fusão entre a membrana interna e a externa do envoltório nuclear.
Mais sobre este tema
editar
A água como solvente e a importância do pH
editarA água, solvente da Vida
editar
A vida, tal como a conhecemos, depende da presença de água. O organismo humano possui cerca de 70% de água, um constituinte fundamental do meio intracelular e de fluidos extracelulares como o sangue. Uma solução em que a água é o único ou principal solvente é denominada solução aquosa.
Nas zonas do planeta em que a água escasseia, os seres vivos possuem adaptações para minimizar a sua perda. Crê-se também que a vida surgiu em meio aquoso, onde os reagentes poderiam circular, encontrar-se e formar ligações, formando moléculas cada vez mais complexas que se agregariam em organismos simples.
A água possui características químicas e físicas muito particulares. Entre elas, o facto de possuir uma densidade menor no estado sólido que no estado líquido, permitindo a flutuação do gelo e a existência de vida subaquática a baixas temperaturas. Também o tipo de ligação química existente entre as moléculas de água, a chamada ligação de hidrogênio, desempenha um papel fundamental em muitos processos biológicos, especialmente em reações catalisadas por diversas enzimas. A compreensão do funcionamento e da função da água em sistemas biológicos é fulcral para o entendimento de processos bioquímicos.
Estrutura da molécula de água
editarA molécula de água é constituída por dois átomos de hidrogênio ligados a um de oxigênio, com uma estrutura angular. O átomo de oxigênio partilha dois dos seus seis elétrons de valência com os átomos de hidrogênio para formar as ligações covalentes entre oxigênio e hidrogênio. Como resultado, o hidrogênio tem a sua camada de valência completa e dedicada à ligação. O átomo de oxigênio possui dois pares de elétrons de valência que não participam então em ligações, mas que produzem uma zona de carga negativa que tende a repelir ligeiramente os átomos de hidrogênio. Por esta razão, a molécula de água não é linear,e sim angular, formando antes um ângulo com aproximadamente 104,5º.

O oxigênio é uma molécula com elevada eletronegatividade, maior que a do hidrogênio, ou seja, tende a atrair mais facilmente elétrons. Embora a ligação covalente seja um tipo de ligação química que exija a partilha eletrônica, é mais provável encontrar esses elétrons mais perto do núcleo do oxigênio que dos núcleos do hidrogênio. Por esta razão, a nuvem eletrônica da molécula de água é mais densa nas imediações do oxigênio, tendo uma carga elétrica local mais negativa e conferindo uma polaridade elétrica à molécula.
Por causa desta polaridade, um átomo de oxigênio pertencendo a uma determinada molécula de água tende a atrair um átomo de hidrogênio de uma molécula vizinha, estabelecendo uma ligação intermolecular denominada ligação de hidrogênio (também é usado o termo ponte de hidrogênio). Este tipo de ligação ocorre entre átomos de hidrogênio e átomos de elevada eletronegatividade, como o já referido oxigênio ou ainda o nitrogênio ou o fósforo, sempre que o hidrogênio tenha uma deficiência eletrônica devida à polarização da molécula em que se encontra (ou seja, sempre que se encontre ligado covalentemente a outro átomo eletronegativo).

Este tipo de ligação tem uma energia relativamente baixa (23 kJ/mol), a suficiente para estabelecer a ligação mas também ser facilmente quebrada. Este é um aspecto importante para a mobilidade das moléculas de água, que estão a associar-se e a dissociar-se constantemente quando no estado líquido, mas que se encontram sempre envolvidas neste tipo de ligação. A água pode ser então pensada como uma rede de moléculas coesas, mas não estáticas como num sólido; esta coesão confere-lhe uma densidade elevada em comparação com outros líquidos à mesma temperatura e causa a existência de uma elevada tensão superficial.
Outras propriedades significativamente afetadas pela existência de ligações de hidrogênio são a temperatura de ebulição e a temperatura de fusão. Este tipo de ligação aumenta estas temperaturas em relação a compostos similares que não a possuam. Por exemplo, o metano, CH4, é um gás a 25º, o que não acontece com o álcool derivado deste, o metanol (CH3OH), um líquido a esta mesma temperatura. A ligação C-H não é muito polar, não havendo grande polarização em moléculas orgânicas que não possuam átomos fortemente eletronegativos.
Solubilidade em água
editarComo a água é um dos constituintes fundamentais da célula, é importante reconhecer que moléculas são solúveis ou não em meio aquoso. As moléculas podem dividir-se em hidrofílicas, ou solúveis em água, e hidrofóbicas, ou insolúveis em água. Em geral, moléculas polares são hidrofílicas e moléculas apolares são hidrofóbicas.
Exemplos de moléculas hidrofóbicas são os ácidos gordos, que formam os fosfolípidos. Estes são constituintes das membranas celulares que possuem uma extremidade ("cabeça") polar e outra ("cauda") apolar. Como consequência, os fosfolípidos são moléculas anfipáticas, ou seja, parcialmente polares e parcialmente apolares. Esta característica possibilita a formação de bicamadas lipídicas que limitam o transporte de moléculas para dentro e fora da célula, separando o meio aquoso interno do externo.
Gases como o O2 e o CO2 são apolares, tendo uma baixa solubilidade em água. Podem, no entanto, permear membranas.
A grande maioria das moléculas orgânicas encontradas em sistemas vivos são hidrofílicas, podendo difundir na célula ou ser transportadas por fluidos dentro de um organismo, associadas ou não a outras moléculas. A maioria dos metabolitos caem nesta categoria.
As proteínas podem ser hidrofílicas ou hidrofóbicas. Quando possuem zonas hidrofóbicas expostas ao solvente, estão normalmente associadas a membranas, podendo ter zonas expostas ao meio aquoso intracelular, extracelular ou a ambos.
A água dissolve sais como o NaCl, dissociando-os nos seus íons; estes não se reassociam porque a água solvata-os de forma eficaz, criando uma barreira física à sua reassociação. Também grupos ionizáveis como o carboxilo, -COOH, presentes em diversas moléculas biológicas, têm a sua forma ionizada (neste caso, -COO-) estabilizada em solução.
Autoionização da água
editarA água sofre autoionização, ionizando-se a H+ e OH-. Na realidade, o próton, H+, não tem existência própria: é capturado por uma molécula de água, originando o ion H3O+ (íon hidrônio ou hidroxônio). Esta ionização é expressa como um equilíbrio químico:

- 2 H2O H3O+ + OH-

A extensão desta ionização é bastante pequena: ambos os íons encontram-se a uma concentração cerca de 10-7 M.L-1.
A capacidade da água em ionizar-se tem consequências de grande relevância fisiológica. Diversas reações bioquímicas dependem da transferência de H+ entre moléculas e enzimas, e a transferência de prótons através das redes formadas por moléculas de água é possibilitada pelo seu pequeno tamanho.
A existência de espécies químicas com possibilidade de se ionizarem em solução altera o equilíbrio da reação de autoionização da água. A extensão do equilíbrio das espécies iônicas H3O+ e OH- é expresso como um normal equilíbrio químico, ou seja, como a razão entre o produto das concentrações dos íons e o produto dos reagentes.
![{\displaystyle K_{eq}={\frac {[H^{+}][OH^{-}]}{[H_{2}O]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/a486b0f937474d27c790ad88d464a92a477b0f46)
Sendo o solvente também o único reagente, a concentração da água numa solução aquosa é constante: a 25ºC, é igual a 55,5 M. A expressão de equilíbrio pode ser rearranjada da seguinte forma:
![{\displaystyle K_{eq}={\frac {[H^{+}][OH^{-}]}{55,5M}}\Leftrightarrow {(55,5M)(K_{eq})}={[H^{+}][OH^{-}]}\Leftrightarrow {K_{w}=[H^{+}][OH^{-}]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/f8f086e4d4384490eccc3d95108149d5add1a531)
em que Kw é designado produto iônico da água. O valor de Keq é também conhecido (1,8×10-16M), pelo que
![{\displaystyle K_{w}=[H^{+}][OH^{-}]=1,0\times 10^{-14}M^{-2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/9e238e2b0ccd4c50f4d22b1577d55ea7cd5c05b6)
O conceito de pH
editarQuando [H+]=[OH-], a concentração de cada uma destas espécies é 1,0×10-7M, a 25ºC. Nestas condições diz-se que a solução se encontra a pH neutro.
O pH é definido como o inverso do logaritmo da concentração de H+:
![{\displaystyle pH=-log[H^{+}]=log{\frac {1}{[H^{+}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e16d6fd265cb3d747505b621f566ccfe66aebf81)
Por esta definição, o pH neutro define-se como sendo numericamente igual a 7 (sem unidade). Quando [H+]<[OH-], a solução terá um pH superior a 7 e diz-se que é básica ou alcalina. Quando [H+]>[OH-], a solução tem um pH inferior a 7, dizendo-se que é uma solução ácida.
Pela definição dada acima, é possível estabelecer uma escala numérica de pH que vai de 1 a 14. Denotar que quando o pH sobe de um valor, na realidade a solução de pH maior é dez vezes mais básica, devido à natureza logarítmica da escala. Dois valores de diferença correspondem a uma diferença de cem vezes, três valores a mil vezes, etc.
De referir que também é possível estabelecer uma escala de pOH, de forma similar à de pH. No entanto, esta não é vulgarmente usada porque em processos biológicos refere-se normalmente a presença ou ausência de prótons, sendo a escala de pH mais prática para o efeito.

Que importância tem o pH de uma solução? Muitas substâncias possuem grupos que podem sofrer protonação, isto é, incorporar um ou mais prótons; da mesma forma, podem sofrer desprotonação, ou seja perder prótons. Em muitos casos, o estado de protonação de uma molécula afeta a sua atividade biológica. Exemplo disto é o estado de protonação de diversas cadeias laterais de aminoácidos que constituem enzimas: por vezes, basta um aminoácido não possuir um próton para uma enzima inteira não funcionar.

O pH de uma solução pode ser medido de várias formas. O método de maior sensibilidade é o uso de um eletrodo de pH, um dispositivo eletroquímico que mede a concentração de H+ em solução. O eletrodo é parcialmente submergido na solução a medir; produz então uma corrente elétrica proporcional à concentração de H+, que é convertida a um valor numérico. Para leituras de menor sensibilidade, podem usar-se fitas de pH ou soluções indicadoras. As soluções indicadoras mudam de cor no chamado ponto de viragem, tendo uma determinada cor abaixo desse valor de pH e outra acima. As fitas de pH usam o mesmo princípio mas em geral usam combinações de indicadores para uma medição mais precisa do pH.
A constante de ionização
editarQualquer solução aquosa tem um determinado valor de pH. Não só a água tem capacidade de se ionizar: muitas substâncias ionizam-se em solução aquosa. Como tal, também podem ser divididas em ácidos, se provocam o abaixamento de pH da solução, e bases, se aumentam o pH.
Neste contexto, um ácido pode ser definido simplesmente como uma substância que doa prótons, enquanto uma base é uma aceitadora de prótons. Um ácido que perde os seus prótons torna-se numa base, enquanto uma base que ganha prótons passa a ser por definição um ácido. Tais pares são denominados pares conjugados ácido/base.
Quando um ácido é forte, dissocia-se totalmente em solução. Se o ácido for representado como HA, a sua dissociação é representada pela equação química
- HA H+ + A-

em que A- é a base conjugada de HA. Em Bioquímica, estes ácidos têm pouco interesse porque não são usuais em sistemas biológicos. São no entanto mais usuais os ácidos fracos, ou seja, aqueles que não se dissociam totalmente em solução. Estabelece-se então um equilíbrio químico entre a espécie protonada e a espécie desprotonada; neste caso, a ionização do ácido é representada como:
- HA H+ + A-
Tal como para a autoionização da água, pode definir-se uma constante de equilíbrio para a ionização de um ácido. Neste contexto, a constante é denominada constante de acidez, Ka.
![{\displaystyle K_{a}={\frac {[H^{+}][A^{-}]}{[HA]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/83ee86c6746a584bd7b209324db405b0563af917)
Um exemplo comum de ácido fraco é o ácido acético, CH3COOH, que se ioniza a ânion acetato e doa um próton nesse processo:
- CH3COOH H+ + CH3COO-
cuja respectiva constante de acidez é então definida por:
![{\displaystyle K_{a}={\frac {[H^{+}][CH_{3}COO^{-}]}{[CH_{3}COOH]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/89d0a720f13720bcafffcaaa80b2c25c7ed39ea4)
Neste caso, é o grupo carboxilo, -COOH, que sofre ionização. Este é um dos grupos encontrados em diversas moléculas biológicas cujas propriedades acídicas são importantes de reconhecer.
Quanto mais forte é um ácido, mais este se dissocia em solução aquosa e maior é Ka. O caso demonstrado assume que o ácido é monoprótico, ou seja, que doa apenas um próton. Este não é sempre o caso, existindo ácidos dipróticos (que doam dois prótons) e tripróticos (doam três prótons). Os ácidos que doam mais de um próton têm constantes de acidez específicas para cada ionização: a primeira ionização tem um Ka1 relativamente baixo, a segunda ionização um Ka2 um pouco maior e a terceira (a haver) tem o Ka3 mais elevado.
Da mesma forma como se definiu pH, é possível definir o pKa de um ácido, sendo o logaritmo do inverso de Ka:

Titulação e zona tampão de um par conjugado ácido/base
editarO pKa é uma grandeza que permite saber a força de um ácido de forma mais intuitiva que através do valor de Ka. Quanto menor é o pKa de um ácido, maior é a sua tendência a ionizar-se e, consequentemente, mais forte é o ácido.
Quando o pH de uma solução aquosa contendo um ácido fraco é igual ao pKa desse ácido, [HA]=[A-]. O valor de pKa de um ácido pode ser facilmente calculado através de uma curva de titulação.
Uma titulação consiste na adição de uma base a uma solução aquosa de um ácido (ou um ácido a uma solução aquosa de uma base) em pequenas quantidades, medindo-se o pH da solução após cada adição. A solução que se adiciona é chamada titulante e a que sofre a adição é titulada. O resultado de tais medições (título) é uma curva de forma sinusoidal em que o centro geométrico corresponde ao pKa do ácido. Num contexto bioquímico, interessa saber as propriedades de ácidos e bases fracos, pelo que as titulações destes são sempre feitas com bases e ácidos fortes, respectivamente.

Por que tem a curva de titulação uma forma sinusoidal? Considere-se que numa solução aquosa de um ácido fraco têm-se dois equilíbrios químicos, nomeadamente o da autoionização da água e a dissociação do ácido:
- 2 H2O H3O+ + OH-
- 2 HA H+ + A-
Esta solução terá um dado pH, de valor relativamente baixo, pois o ácido encontra-se ligeiramente dissociado. Ao adicionar-se uma base forte como por exemplo o hidróxido de sódio (NaOH), esta dissocia-se totalmente em solução, havendo então ânions OH- que podem combinar-se com o H+ "livre", formando H2O. Esta reação força a diminuição da concentração de H+ em solução; pelo princípio de Le Chatelier, o ácido terá de se dissociar um pouco mais para compensar esta "falta" de H+, atingindo-se novo equilíbrio entre ácido e a sua base conjugada. À medida que se acrescenta mais OH-, cada vez mais ácido se dissocia. Chega-se então a um ponto em que metade de HA que existia no início encontra-se sob a forma da sua base conjugada, A-: está-se a meio da titulação, forma adicionados 0,5 equivalentes de OH- ao ácido e o pH neste ponto é igual ao pKa do ácido.
À medida que se acrescenta mais OH-, o pH continua a subir e o ácido a dissociar-se. No fim da titulação, todo o ácido encontra-se dissociado sob a forma de A-.
Como se pode notar, na zona central do gráfico a variação de pH aumenta com a adição de quantidades equivalentes de base. Esta zona é chamada zona tampão e tem grande importância biológica, como será evidente mais adiante. A zona tampão existe porque durante essa parte da titulação existem dois equilíbrios (o da autoionização da água e o da dissociação do ácido) que se compensam mutuamente, fazendo com que a concentração de H+ não se altere significativamente.
É possível relacionar a concentração do tampão com o pH e o pKa através da chamada equação de Henderson-Hasselbalch. Partindo-se da definição de constante de acidez e pondo em evidência a concentração de H+, pode rearranjar-se a relação sob a forma:
![{\displaystyle [H^{+}]=K_{a}{\frac {[HA]}{[A^{-}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/833ae0a24a2de71e428b92ff18b645d006564329)
Se se aplicar o logaritmo negativo a ambos os termos da equação, tem-se:
![{\displaystyle -log[H^{+}]=-logK_{a}-log{\frac {[HA]}{[A^{-}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/b3a0d22a313f75bb8b58752e52cad6528e4152eb)
Como -logX = pX, a equação pode ser escrita da forma:
![{\displaystyle pH=pK_{a}-log{\frac {[HA]}{[A^{-}]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e05e8234f65644b3ce501e99c5a965196b513208)
ou ainda
![{\displaystyle pH=pK_{a}+log{\frac {[A^{-}]}{[HA]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7ce0e4959195eb3a7fc22e9215f917e7b7c6c8a7)
sendo esta a forma mais conhecida da equação de Henderson-Hasselbalch. Esta relação matemática explica a forma quasi-sinusoidal da curva de titulação e mostra também que pH = pKa quando as concentrações de ácido e da sua base conjugada são iguais.
As soluções tampão em sistemas biológicos
editarUma solução tampão é uma solução aquosa de um ácido e da sua base conjugada que não sofre variações significativas de pH quando se adicionam pequenas quantidades de ácidos ou bases. São portanto soluções cujo pH ideal se encontra no centro da zona tampão do par conjugado ácido/base. Como já referido, muitos processos biológicos dependem do estado de protonação de moléculas como as enzimas, sendo portanto fundamental o controle rigoroso do pH do meio em que esses processos se desenrolam. Fluidos como o sangue e o citoplasma têm um pH definido, geralmente em torno de 7, e que não muda significativamente graças à presença de diversas substâncias dissolvidas que atuam como tampão.
O citoplasma é rico em proteínas; os grupos laterais ionizáveis de aminoácidos que constituem essas proteínas têm um papel fundamental no tamponamento do meio intracelular. Outras moléculas ionizáveis, como o ATP, ácidos nucleicos e compostos intermediários de vias metabólicas, entre outros, contribuem também para a manutenção de um valor mais ou menos estável de pH no interior da célula.
Dois dos tampões fisiológicos mais importantes, especialmente em fluidos como o sangue, são o tampão de carbonatos e o tampão de fosfatos. O dióxido de carbono (CO2), um gás em condições normais de pressão e temperatura, pode dissolver-se em soluções aquosas formando ácido carbônico, H2CO3. Estabelece-se então o equilíbrio
- CO2 (g) + H2O (l) H2CO3 (aq)
Por sua vez, o ácido carbônico dissocia-se em solução aquosa, estabelecendo-se um segundo equilíbrio:
- H2CO3 HCO3- + H+
A quantidade de íon hidrogenocarbonato (HCO3-) em solução depende em primeira instância da pressão parcial do CO2, pois esta determina o equilíbrio entre CO2 dissolvido e não dissolvido em solução. Assim, quanto mais dióxido de carbono for dissolvido, maior será a acidificação da solução aquosa em que este se dissolve.
O tampão de fosfatos, em situação fisiológica, refere-se especificamente ao equilíbrio
- H2PO4- HPO42- + H+
sendo um tampão natural no citoplasma de todas as células, já que o grupo fosfato está presente em diversas moléculas biológicas.
Em trabalho laboratorial biológico e bioquímico é comum trabalhar-se com soluções tampão para manter o material de estudo (células, enzimas, tecidos, etc.) num meio com pH definido. Diversas soluções tampão podem ser feitas, dependendo do pH a que se pretende manter o material. Para isto. é necessário saber qual o pH ótimo desse material, ou seja, o pH a que se pode detectar um máximo de atividade fisiológica. Esta atividade está relacionada não só com o estado de protonação dos metabolitos envolvidos em reações bioquímicas, mas também (e principalmente) com a estabilidade e estado de protonação das enzimas envolvidas nessas reações. Os tampões de carbonatos e fosfatos são vulgarmente usados, mas outros de origem sintética, como o Tris-HCl (base Tris(hidroxilamina)aminometano, ácido HCl) ou o HEPES (ácido N-(2-hidroxietilo)-piperazina-N'-2-etanesulfónico, uma molécula anfotérica), são também de uso comum.
Esta página foi eleita pelos colaboradores como uma das melhores do Wikilivros. Para mais informações, consulte a página de votações.

Aminoácidos e Proteínas
editar| Este módulo precisa ser revisado por alguém que conheça o assunto (discuta). |
Aminoácidos
editar
Os aminoácidos são moléculas que contêm simultaneamente grupos funcionais amina e ácido carboxílico. Em Bioquímica, este termo é usado como termo curto e geral para referir os aminoácidos alfa, ou seja, aqueles em que as funções amino e carboxilato estão ligadas a um carbono alifático, denominado carbono-alfa (carbono-α). Pelo menos um átomo de hidrogênio está ligado a este carbono. A esfera de coordenação do carbono-α é completada com a presença de uma cadeia lateral, diferente para diferentes aminoácidos. Os aminoácidos α (cerca de vinte) são constituintes de todas as proteínas e peptídeos.
Em soluções aquosas de pH neutro, os aminoácidos podem existir em duas formas. Uma pequena fração encontrar-se-á numa forma eletricamente neutra, ou seja, com o grupo amina desprotonado (-NH2) e o grupo carboxilo protonado (-COOH). A maioria estará, no entanto, numa forma ionizada, em que o grupo amina se encontra protonado (-NH3+) e o ácido carboxílico desprotonado a carboxilato (-COO-), denominando-se esta forma de zwitteriónica (do alemão zwitter, que significa "híbrido"). Um zwitterião é uma molécula globalmente neutra em termos de carga elétrica mas possuindo cargas locais devido à presença de grupos ionizados.
Os aminoácidos podem ligar-se entre si com uma ligação amida, que em Bioquímica é especificamente designada, neste caso, de ligação peptídica. A ligação ocorre entre o átomo de carbono do grupo carboxilato e o nitrogênio do grupo amina; no processo, é libertada uma molécula de água, seno a ligação final entre o carbono de um grupo carbonilo e o nitrogênio de uma amina secundária. Como consequência, uma cadeia peptídica, ou seja, formada por diversos aminoácidos ligados desta forma, terá um grupo amina numa extremidade (denominada N-terminal) e um grupo carboxilato na extremidade oposta (denominada C-terminal).
A ligação peptídica tem uma geometria planar porque existe ressonância entre o grupo carbonilo e o nitrogênio da amina, fazendo com que a ligação C-N tenha um caráter parcial de ligação dupla (é possível desenhar uma estrutura de ressonância entre o átomo de carbono e o de nitrogênio, tendo uma carga negativa formal sobre o oxigênio e uma positiva sobre o nitrogênio). Esta característica impede que haja rotação em torno da ligação C-N, que se mantém numa conformação trans. Seis átomos encontram-se então no mesmo plano geométrico: o carbono-α de um aminoácido, os átomos do grupo carbonilo da ligação peptídica, os átomos da amina secundária dessa mesma ligação e o carbono-α do segundo aminoácido.
Simbologia e nomenclatura
editarNa nomenclatura dos aminoácidos, a numeração dos carbonos da cadeia principal é iniciada a partir do carbono do grupo carboxilato.
| Nome vulgar | Símbolo (3 letras) | Símbolo (1 letra) | Nome sistemático |
|---|---|---|---|
| Glicina ou Glicocola | Gly | G | Ácido 2-aminoacético ou ácido 2-amino-etanóico |
| Alanina | Ala | A | Ácido 2-aminopropiónico ou ácido 2-amino-propanóico |
| Leucina | Leu | L | Ácido 2-aminoisocapróico ou ácido 2-amino-4-metil-pentanóico |
| Valina | Val | V | Ácido 2-aminovalérico ou ácido 2-amino-3-metil-butanóico |
| Isoleucina | Ile | I | Ácido 2-amino-3-metil-n-valérico ou ácido 2-amino-3-metil-pentanóico |
| Prolina | Pro | P | Ácido pirrolidino-2-carboxílico |
| Fenilalanina | Phe | F | Ácido 2-amino-3-fenil-propiônico ou ácido 2-amino-3-fenil-propanóico |
| Serina | Ser | S | Ácido 2-amino-3-hidroxi-propiónico ou ácido 2-amino-3-hidroxi-propanóico |
| Treonina | Thr | T | Ácido 2-amino-3-hidroxi-n-butírico |
| Cisteína | Cys | C | Ácido 2-bis-(2-amino-propiónico)-3-dissulfeto ou ácido 3-tiol-2-amino-propanóico |
| Tirosina | Tyr | Y | Ácido 2-amino-3-(p-hidroxifenil)propiónico ou paraidroxifenilalanina |
| Asparagina | Asn | N | Ácido 2-aminossuccionâmico |
| Glutamina | Gln | Q | Ácido 2-aminoglutarâmico |
| Aspartato ou ácido aspártico | Asp | D | Ácido 2-aminossuccínico ou ácido 2-amino-butanodióico |
| Glutamato ou ácido glutâmico | Glu | E | Ácido 2-aminoglutárico |
| Arginina | Arg | R | Ácido 2-amino-4-guanidina-n-valérico |
| Lisina | Lys | K | Ácido 2,6-diaminocapróico ou ácido 2,6-diaminoexanóico |
| Histidina | His | H | Ácido 2-amino-3-imidazolpropiónico |
| Triptofano | Trp | W | Ácido 2-amino-3-indolpropiónico |
| Metionina | Met | M | Ácido 2-amino-3-metiltio-n-butírico |
Ponto isoelétrico
editarO ponto isoelétrico (pI) de uma molécula é o pH ao qual essa molécula é eletricamente neutra. Este conceito é aplicado particularmente a aminoácidos e proteínas. O ponto isoelétrico não é o pH em que todas os grupos básicos estão desprotonados e os ácidos protonados, mas antes o pH em que o número de cargas positivas e negativas da molécula se cancelam a zero.
O pI corresponde à média dos valores de pKa da molécula:
- pI=(pK1+pK2+...+pKn)/n
No caso de aminoácidos isolados com cadeias laterais ionizáveis, o pI do aminoácido será a média dos pKa dos grupos amino e carboxilato ligados ao carbono-α e ainda do pKa da cadeia lateral. As proteínas têm normalmente cadeiasionizáveis, e a diferente proporção de diferentes aminoácidos em cada tipo de proteína confere-lhes diferentes valores de pI.
O cálculo teórico de pI torna-se mais difícil quanto maior for a proteína, já que o pI das cadeias laterais de aminoácidos podem variar ligeiramente dentro do ambiente proteico. É no entanto possível fazer a determinação experimental do pI através de eletroforese em gel de poliacrilamida. A proteína cujo pI se pretende determinar é aplicada num gel ao longo do qual existe variação de pH; ao aplicar-se uma diferença de potencial entre as duas extremidades do gel, a proteína migra através deste até encontrar uma zona de pH igual à do seu pI. Neste ponto, a sua migração para, pois a carga da proteína é neutra a esse pH e não responderá mais à aplicação de um potencial elétrico. Esse valor de pH será então o pI da proteína.
Isomeria
editarTodos os aminoácidos obtidos pela hidrólise de proteínas em condições suficientemente suaves apresentam atividade óptica, à exceção da glicina. Como os aminoácidos apresentam quatro grupos diferentes ligados ao carbono central, os aminoácidos apresentam quiralidade, sendo o carbono-α um centro quiral.
O centro quiral permite a existência de estereoisômeros, devido aos diferentes arranjos espaciais possíveis, apresentando os aminoácidos uma atividade óptica. Mais especificamente, existem diferentes enantiômeros, ou seja, formas de aminoácidos que são a imagem do espelho uma de outra. Os enantiómeros de aminoácidos são usualmente classificados como D ou L, sendo essa classificação referente à semelhança com a estrutura do D-gliceraldeído e do L-gliceraldeído, respectivamente. Somente os L-aminoácidos são constituintes das proteínas.
Síntese
editarTodos os aminoácidos são derivados de intermediários da glicólise, do ciclo dos ácidos tricarboxílicos ou das via das pentoses-fosfato. O nitrogênio entra nessas vias através do glutamato. Há uma grande variação no nível de complexidade das vias, sendo que alguns aminoácidos estão a apenas alguns passos enzimáticos dos seus precursores e em outros as vias são complexas, como no caso dos aminoácidos aromáticos.
As vias biossintéticas de aminoácidos são agrupadas de acordo com a família dos precursores de um deles. As principais famílias são:
- A do α-cetoglutarato, que origina o glutamato, a glutamina, a prolina e a arginina.
- A do 3-fosfoglicerato, de onde são derivados a serina, a glicina e a cisteína.
- A do oxaloacetato, que dá origem ao aspartato, que vai originar a asparagina, a metionina, a treonina e a lisina.
- A do piruvato, que dá origem a alanina, a valina, a leucina e a isoleucina.
Estrutura e classificação dos aminoácidos
editar
Classificação
editarOs aminoácidos podem ser classificados nutricionalmente, quanto à sua cadeia lateral e quanto ao seu destino.
Aminoácidos essenciais e não essenciais
editarUm aminoácido essencial é aquele que o organismo (considerado normalmente, o humano) não é capaz de sintetizar mas é requerido para o seu funcionamento.
O organismo humano é incapaz de sintetizar cerca de metade dos vinte aminoácidos comuns. Tem então de os obter através da dieta, pela ingestão de alimentos ricos em proteínas.
Os aminoácidos não essenciais são também necessários para o funcionamento do organismo, mas podem ser sintetizados in vivo a partir de determinados metabolitos.
Existem aminoácidos que são essenciais apenas em determinadas situações patológicas ou em organismos jovens e em desenvolvimento. A estes convencionou-se a designação "condicionalmente essenciais". Estes aminoácidos são normalmente fonte de divisão entre os cientistas, havendo os que consideram estes como essenciais e os que não os consideram essenciais.
| Não essenciais | Condicionalmente essenciais | Essenciais |
|---|---|---|
| Alanina | Arginina | Histidina |
| Asparagina | Glutamina | Isoleucina |
| Aspartato | Glicina | Leucina |
| Glutamato | Prolina | Lisina |
| Serina | Tirosina | Metionina |
| Cisteína | Fenilalanina | |
| Treonina | ||
| Triptofano | ||
| Valina |
Os aminoácidos não essenciais possuem, em geral, vias de síntese relativamente simples. Por exemplo, o metabolito α-cetoglutarato (intermediário do ciclo dos ácidos tricarboxílicos) é precursor do glutamato, que por sua vez pode dar origem à glutamina, à prolina e à arginina.
Quanto à cadeia lateral
editarA classificação quanto à cadeia lateral (a negrito) pode ser feita em: Aminoácidos Apolares e Polares. Com uma subdivisão dos polares em Aa polares neutros; Aa polares ácidos e Aa polares básicos.
- Aminoácidos apolares
Apresentam grupos químicos de hidrocarbonetos apolares ou hidrocarbonetos modificados, exceto a glicina (que possui um átomo de hidrogénio como cadeia lateral). São hidrófobos.
- Glicina: H-CH(NH2) -COOH
- Alanina: CH3-CH(NH2) -COOH
- Leucina: CH3(CH3)-CH2-CH(NH2)-COOH
- Valina: CH3-CH(CH3)-CH(NH2)-COOH
- Isoleucina: CH3-CH2-CH (CH3)-CH(NH2)-COOH
- Prolina:-CH2-CH2-CH2- ligando o grupo amino ao carbono alfa
- Fenilalanina: C6H5-CH2-CH(NH2)-COOH
- Triptofano: R aromático-CH(NH2)-COOH
- Metionina: CH3-S-CH2-CH2-CH(NH2)-COOH
- Aminoácidos Polares neutros
Apresentam grupos químicos que tendem a formar ligações de hidrogénio.
- Serina: OH-CH2-CH(NH2)-COOH
- Treonina: OH-CH (CH3)-CH(NH2)-COOH
- Cisteina: SH-CH2-CH(NH2)-COOH
- Tirosina: OH-C6H4-CH2-CH(NH2)-COOH
- Asparagina: NH2-CO-CH2-CH(NH2)-COOH
- Glutamina: NH2-CO-CH2-CH2-CH(NH2)-COOH
- Aminoácidos Polares ácidos
Apresentam grupos carboxilato.São hidrófilos.
- Ácido aspártico: HCOO-CH2-CH(NH2)-COOH
- Ácido glutâmico: HCOO-CH2-CH2-CH(NH2)-COOH
- Aminoácidos Polares básicos
Apresentam grupos amino. São hidrófilos.
- Arginina: HN=C(NH2)-NH-CH2-CH2-CH2-CH(NH2)-COOH
- Lisina: NH2-CH2-CH2-CH2-CH2-CH(NH2)-COOH
- Histidina: H-(C3H2N2)-CH2-CH(NH2)-COOH
Quanto ao destino
editarEssa classificação é dada em relação ao destino tomado pelo aminoácido quando o grupo amina é excretado do corpo na forma de ureia (mamíferos), amônia (peixes) e ácido úrico (aves e répteis).
- Destino cetogênico
Quando o álcool restante da quebra dos aminoácidos vai para qualquer fase do ciclo dos ácidos tricarboxílicos sob a forma de acetil-coenzima A ou outra substância.
- Destino glicogênico
Quando o álcool restante da quebra dos aminoácidos vai para a glicólise.
Fórmula estrutural
editar-
 Alanina (Ala/A)
Alanina (Ala/A) -
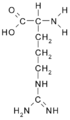 Arginina (Arg/R)
Arginina (Arg/R) -
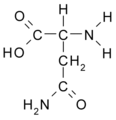 Asparagina (Asn/N)
Asparagina (Asn/N) -
 Ácido aspártico (Asp/D)
Ácido aspártico (Asp/D) -
 Cisteina (Cys/C)
Cisteina (Cys/C) -
 Ácido glutâmico (Glu/E)
Ácido glutâmico (Glu/E) -
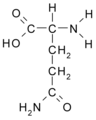 Glutamina (Gln/Q)
Glutamina (Gln/Q) -
 Glicina (Gly/G)
Glicina (Gly/G) -
 Histidina (His/H)
Histidina (His/H) -
 Isoleucina (Ile/I)
Isoleucina (Ile/I) -
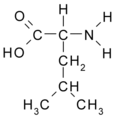 Leucina (Leu/L)
Leucina (Leu/L) -
 Lisina (Lys/K)
Lisina (Lys/K) -
 Metionina (Met/M)
Metionina (Met/M) -
 Fenilalanina (Phe/F)
Fenilalanina (Phe/F) -
 Prolina (Pro/P)
Prolina (Pro/P) -
 Serina (Ser/S)
Serina (Ser/S) -
 Treonina (Thr/T)
Treonina (Thr/T) -
 Triptofano (Trp/W)
Triptofano (Trp/W) -
 Tirosina (Tyr/Y)
Tirosina (Tyr/Y) -
 Valina (Val/V)
Valina (Val/V)




















Estrutura tridimensional
editarAminoácidos apolares
editar-
 Glicina (Gly/G)
Glicina (Gly/G) -
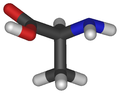 Alanina (Ala/A)
Alanina (Ala/A) -
 Leucina (Leu/L)
Leucina (Leu/L) -
 Isoleucina (Ile/I)
Isoleucina (Ile/I) -
 Valina (Val/V)
Valina (Val/V) -
 Metionina (Met/M)
Metionina (Met/M) -
 Prolina (Pro/P)
Prolina (Pro/P) -
 Fenilalanina (Phe/Fen)
Fenilalanina (Phe/Fen) -
 Triptofano (Trp/W)
Triptofano (Trp/W)









Aminoácidos polares neutros
editar-
 Asparagina (Asn/N)
Asparagina (Asn/N) -
 Cisteína (Cys/C)
Cisteína (Cys/C) -
 Glutamina (Gln/Q)
Glutamina (Gln/Q) -
 Serina (Ser/S)
Serina (Ser/S) -
 Treonina (Thr/T)
Treonina (Thr/T) -
 Tirosina (Tyr/y)
Tirosina (Tyr/y)






Aminoácidos polares ácidos
editar-
 Ácido aspártico (Asp/D)
Ácido aspártico (Asp/D) -
 Ácido glutâmico (Glu/E)
Ácido glutâmico (Glu/E)


Aminoácidos polares básicos
editar-
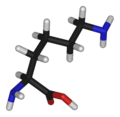 Lihsina (Lys/K)
Lihsina (Lys/K)

Proteínas
editarAs proteínas são um dos constituintes básicos dos organismos, sendo uma das classes de moléculas mais estudadas em Bioquímica. As proteínas fazem parte de maquinaria celular responsável pelo funcionamento da célula. Muitas proteínas são enzimas, ou seja, têm capacidade de catalisar reações bioquímicas. Outras têm um papel estrutural ou mecânico, como as que fazem parte do citoesqueleto ou de poros em membranas. As proteínas são polímeros (cadeias) não ramificados de aminoácidos ligados entre si por ligações peptídicas, podendo ser constituídas por um ou mais de tais polímeros.
O papel e a importância das proteínas
editar
As proteínas são uma classe fundamental de moléculas em Biologia. Estão presentes em todas as formas de vida na Terra, sendo responsáveis pela maioria dos processos mais complexos que tornam a vida possível. e são o principal constituinte estrutural dos seres vivos. De acordo com o dogma central da Biologia Molecular, proposto em 1958 por Francis Crick, a informação hereditária, contida no DNA, é passada de DNA para o RNA e deste para as proteínas. Assim, o DNA é responsável por armazenar a informação necessária para a síntese de proteínas e o RNA toma o papel de transferir essa informação do DNA para a maquinaria de tradução nos ribossomas, onde ocorre a montagem das cadeias polipeptídicas.
Praticamente todos as complexas reações químicas que ocorrem em sistemas vivos são catalisadas por proteínas denominadas enzimas. Como catalizadoras que são, as enzimas aumentam a velocidade de reações químicas sem alterar o seu equilíbrio, possibilitando a existência de reações na célula que de outra forma seriam demasiado lentas para sustentar processos biológicos. Um grupo de moléculas biológicas, as ribozimas, possuem também alguma capacidade catalítica, mas não são constituídas por proteína, sendo antes moléculas de RNA. A maioria do esqueleto que sustenta e mantém a integridade celular é constituído por proteínas.
Existem proteínas envolvidas na transmissão e transdução de sinal do ambiente extracelular para o interior da célula, proteínas que assistem na duplicação do material genético, proteínas envolvidas na transformação da energia da luz em energia química e desta em energia mecânica e proteínas que transportam moléculas entre compartimentos celulares, entre células e até entre diferentes partes de um organismo. São também importantes reservas de azoto nos organismos, estando todo o metabolismo do azoto ligado ao metabolismo dos aminoácidos.
Por outro lado, as proteínas não são capazes de se replicar de forma autônoma, nem são boas reservas de energia química (este papel é desenrolado pelos glicídios e lipídios). Embora façam parte de estruturas como membranas, as proteínas não são por si só capazes das funções de uma membrana lipídica. São capazes, no entanto, de formar uma capa proteica em vírus.
As proteínas como polímeros de aminoácidos
editarAs proteínas são compostas por um ou mais polímeros lineares de aminoácidos ligados entre si por ligações peptídicas. Este tipo de ligação amida resulta da reação de condensação entre um grupo carboxílico alfa de um aminoácido e o grupo amina alfa de outro aminoácido (estes grupos estão ligados ao carbono-α dos respectivos aminoácidos). A cada cadeia pode chamar-se também de péptido. Polímeros de pequenas dimensões (tipicamente com menos de vinte aminoácidos) são denominados oligopeptídios; em geral, uma cadeia simples mais ou menos longa de aminoácidos é denominada polipéptido, pelo que as designações "proteína" e "polipéptido" são muitas vezes usadas sem diferenciação.
Por serem cadeias não ramificadas, os polipéptidos têm numa extremidade um grupo amina que não se encontra envolvido numa ligação peptídica e na outra extremidade um carboxilato nas mesmas condições. A primeira extremidade é então denominada N-terminal e a segunda C-terminal. A sequência pela qual se encontram ligados os aminoácidos é denominada estrutura primária da proteína, mas é mais vulgarmente conhecida apenas por sequência de aminoácidos. Por convenção, estes são numerados começando no N-terminal, o que reflecte a forma como os polipéptidos são sintetizados na célula (também começando no N-terminal).
Como os aminoácidos perdem alguns átomos aquando da formação da ligação peptídica, é usual denominar estes de resíduos de aminoácidos (ou simplesmente resíduos) desde o momento em que fazem parte de uma cadeia polipeptídica.
As cadeias laterais dos aminoácidos são quimicamente muito variáveis, podendo ser polares ou apolares, ionizáveis ou não, tendo diversos tamanhos e níveis de complexidade. Os milhões de possibilidades de combinação de diferentes aminoácidos que uma proteína pode ter explica a complexidade e versatilidade das proteínas em geral.
Diferentes níveis estruturais
editar
As proteínas possuem diferentes tipos de estrutura, além da já mencionada estrutura primária. A sequência de aminoácidos pode organizar-se espacialmente em domínios, sendo esta organização denominada estrutura secundária. Os principais tipos de estrutura secundária são hélices alfa e folhas beta; além destas podem referir-se os random coils (zonas desordenadas) e as beta turn (ligações entre folhas beta).
As hélices alfa são troços de polipéptido com uma forma em hélice em que as cadeia de aminoácidos apontam para o exterior dessa hélice. Este tipo de estrutura é estabilizado pela existência de múltiplas ligações de hidrogénio no interior da hélice. Uma concentração relativamente alta de glicinas no polipéptido tende a forçar a existência de hélices alfa.
A estrutura em folha beta é formada por sequências do polipéptido que se empilham em camadas, havendo uma estabilização desta estrutura também através de ligações de hidrogénio. As folhas podem ter uma conformação em paralelo se se encontrarem na mesma direcção N-terminal—C-terminal ou em antiparalelo se se empilharem em sentidos opostos. As beta turns ligam duas folhas beta com quatro aminoácidos numa conformação definida.
Um random coil é uma zona da proteína que não tem uma estrutura secundária definida.
As proteínas adquirem a sua estrutura final (enrolamento ou folding, a estrutura terciária) de forma espontânea de modo a adquirir uma configuração de energia mínima. In vivo, existem algumas proteínas (denominadas "chaperonas") que ajudam neste enrolamento nalguns casos, em especial quando uma proteína é muito complexa e tende a tomar enrolamentos errados; no entanto, a maioria das proteínas enrola-se de forma correta espontaneamente. É a estrutura primária da proteína que determina o seu enrolamento final. Este enrolamento pode demorar alguns milissegundos.
Devido à enorme complexidade provocada pela existência de inúmeros aminoácidos de natureza química diversa, é difícil prever como uma proteína se enrolará. Existem curtas sequências de aminoácidos que se repetem em diferentes proteínas e que são ou podem ser conhecidos estruturalmente, podendo prever-se como se encontrarão noutras proteínas; estas sequências são denominadas motivos. Um dos maiores problemas em Biologia Molecular é saber se uma proteína pode ser produzida com um enrolamento correto ou não.
As proteínas podem perder a sua estrutura se postas em condições químicas adversas, como pH extremos, ambientes hidrofóbicos, altas concentrações de sais ou altas temperaturas. Este processo é denominado desnaturação. Uma proteína desnaturada não tem uma estrutura definida e tende a agregar-se em massas insolúveis, especialmente encontrando-se numa concentração relativamente elevada. Um exemplo comum de desnaturação ocorre quando um ovo é cozinhado: o branqueamento e solidificação da clara do ovo é devido à desnaturação de proteínas (em particular da albumina) nela contida. Neste caso, o processo é irreversível, ou seja, as proteínas não voltam à sua configuração inicial, mas nem sempre este é o caso: muitas proteínas renaturam quando colocadas num ambiente adequado à formação da sua estrutura.
Pode também falar-se em estrutura quaternária de uma proteína quando se refere à presença de múltiplas cadeias polipeptídicas numa só proteína. Neste caso, diversos (dois ou mais) polipéptidos enrolam-se formando uma proteína. O enrolamento de mais de uma cadeia numa estrutura é estabilizado pela presença de ligações químicas intermoleculares, em particular ligações dissulfureto, que ligam as diferentes cadeias numa só unidade.
A importância da estrutura das proteínas
editarDe uma forma geral, a função de uma proteína é determinada pela sua estrutura. Macromoléculas como o DNA, que não efetuam muitas funções diferentes, têm pouca diversidade estrutural. As proteínas têm uma diversidade estrutural muito maior, que se reflete nas múltiplas funções que assumem em sistemas vivos. A manutenção de uma estrutura é vital para o funcionamento correto da proteína, existindo por isso situações patológicas associadas a maus enrolamentos da estrutura de determinadas proteínas. As enzimas têm de ter uma conformação espacial tal que possam reconhecer o seu substrato, havendo necessidade de uma complementaridade espacial para uma eficiente catálise. Proteínas estruturais como o colágeno têm de sofrer stress mecânico e ainda assim serem capazes de manter uma matriz regular onde as células podem aderir e proliferar. Proteínas com funções motoras têm de ser capazes de converter energia química em movimento de uma forma precisa, como no caso de poros membranares que regulam a entrada e saída de íons da célula.
Modificações pós-traducionais
editarMuitas proteínas sofrem modificações após a sua síntese dentro da célula. Estas são provocadas por modificações químicas nas cadeias laterais de alguns resíduos de aminoácidos. As mais comuns são:
- oxidação,
- acilação,
- glicosilação,
- metilação.

A modificação de cadeias laterais pode conferir propriedades específicas a proteínas tais como a capacidade de reconhecer outras moléculas ou de se integrar na membrana plasmática.
Ligações dissulfureto
editarAs ligações dissulfureto formam-se entre os átomos de enxofre de dois resíduos de cisteína. Esta ligação resulta da oxidação dos grupos sulfidrilo das cisteínas, que resulta numa ligação covalente entre os átomos de enxofre (-S-S-). Estas ligações dificilmente se formam na superfície e uma proteína porque existem muitas substâncias de natureza redutora no citoplasma. São importantes na manutenção da estrutura de uma proteína, podendo ocorrer entre cisteínas de uma mesma ou de diferentes cadeias polipeptídicas.
Em algumas enzimas, a formação e redução de ligações dissulfureto tem grande importância na sua atividade biológica.
Enzimas
editar
Para que a vida seja possível, é necessário que as reações químicas que a sustentam se deem a uma determinada velocidade. Muitas das reações bioquímicas não se realizariam a uma velocidade relativamente elevada se não fossem catalisadas. Em sistemas vivos, a catálise de reações químicas é feita por enzimas.
As enzimas são proteínas que, atuando como catalisadores na maioria das reações bioquímicas, baixam a energia de ativação necessária para que se dê uma reação química. Por serem catalisadores eficientes, são aproveitadas para aplicações industriais, como na indústria farmacêutica ou na alimentar. Como intervêm nas reações químicas que sustentam a vida, a compreensão do seu funcionamento é importante em áreas como a investigação de patologias com origem em deficiências enzimáticas.
A esmagadora maioria das reações bioquímicas dá-se em vias metabólicas, que são sequências de reações em que o produto de uma reação é utilizado como reagente na reação seguinte. Diferentes enzimas catalisam diferentes passos de vias metabólicas, agindo de forma concentrada de modo a não interromper o fluxo nessas vias. Por outro lado, como será explicado mais adiante, cada enzima pode sofrer regulação da sua atividade, aumentando-a, diminuindo-a ou mesmo interrompendo-a. Assim, o fluxo de uma via metabólica depende da velocidade de catálise das enzimas que nela participam.
Um pequeno grupo de enzimas, as ribozimas, têm uma natureza não-proteica. As ribozimas são moléculas de RNA que possuem capacidade catalítica. Este grupo de enzimas, que possui propriedades peculiares, será abordado à parte das enzimas proteicas.
Estrutura
editar
Estruturalmente, as enzimas possuem todas as características das proteínas, tendo zonas da sua estrutura responsáveis pela catálise. A zona reativa da enzima é denominada centro ativo e é onde se liga o reagente (substrato) que vai ser transformado no produto. Podem existir também outras zonas da cadeia polipeptídica que são sensíveis à presença de determinadas espécies químicas, modulando a atividade da enzima. tais zonas são denominadas centros alostéricos e essa modulação de alosteria.
A manutenção da estrutura de uma enzima é de particular importância para a sua atividade: esta pode ser perdida se a enzima é colocada num meio em que fatores como o pH ou a temperatura não favoreçam a estabilidade estrutural da cadeia polipeptídica.
Algumas enzimas necessitam da presença de outras espécies químicas, genericamente denominadas cofatores, para efetuar a catálise. A natureza química dos cofatores é muito diversa: podem ser íons metálicos, como o Mg2+, o Zn+ ou o Fe2+, moléculas orgânicas, como o fosfato de piridoxal ou a coenzima A, e ainda moléculas orgânicas contendo metais, como o grupo hemo (uma porfirina contendo ferro) ou a vitamina B12 (5'-desoxiadenosilcobalamina).
Dois termos relacionados com cofatores que alguns autores tendem a deixar cair em desuso mas que são ainda frequentemente encontrados são:
- coenzima: refere-se a cofatores complexos, que não são apenas íons metálicos;
- grupo prostético: cofator ligado de forma covalente à cadeia polipeptídica.
Mecanismo
editarAs enzimas atuam diminuindo a energia de ativação da reação que catalisam, não alterando, no entanto, o seu equilíbrio. Em geral, uma enzima catalisa apenas um substrato, algo que é condicionado pela estrutura do centro ativo da enzima. Este possui uma geometria definida e determinadas características físico-químicas (hidrofobicidade, carga elétrica local) que condicionam o tipo de substrato que pode acessar, ligar-se e sofrer alteração química no centro ativo.
Quando um substrato se liga ao centro ativo, forma-se o chamado complexo enzima-substrato (ES). Embora esta designação possa parecer uma formalidade, a formação do ES é importante na determinação da velocidade de reação e, por conseguinte, na velocidade de formação de produto(s). O substrato sofre uma alteração enquanto se encontra ligado à enzima, transformando-se num produto; existe então, de forma transiente, um complexo enzima-produto. O produto desliga-se posteriormente da enzima e esta encontra-se preparada para novo ciclo catalítico.
Propriedades
editarPor serem proteínas, cada enzima possui um pH ótimo e uma temperatura ótima de funcionamento. Acima dessa temperatura a enzima começa a sofrer desnaturação, perdendo a sua estrutura tridimensional e por conseguinte a sua capacidade catalítica. A temperaturas baixas as enzimas encontram-se "adormecidas", pois há uma diminuição drástica da atividade da enzima, no entanto a partir de uma temperatura mínima a velocidade de reação aumenta até um valor ótimo, que, na maioria das vezes, é cerca de 40ºC.
Apesar de serem sintetizadas in vivo, as enzimas podem funcionar fora da célula (in vitro), possibilitando o seu estudo funcional e estrutural. Podem também ser imobilizadas num suporte sintético para uso industrial (por exemplo, em biorreatores usados para limpeza de efluentes) ou laboratorial (por exemplo, em eletrodos que detectam glicose, usando a enzima glicose oxidase).
Por serem catalisadores, uma mesma enzima pode catalisar várias vezes a mesma reação, algo que o fazem muito rapidamente (milhares de vezes por segundo). No entanto, pela mesma razão, as enzimas não alteram o equilíbrio químico da reação que catalisam.
As enzimas são específicas para o seu substrato, catalisando uma só reação. Existem algumas enzimas que catalisam substratos similares, mas normalmente são mais específicas para um deles.
Nomenclatura
editarÀ medida que enzimas foram sendo descobertas, receberam nomes arbitrários. Como exemplo, a lisozima recebeu o seu nome por ter a capacidade de fazer a lise (ruptura) da parede celular de determinadas bactérias. Tendo nascido a necessidade de sistematizar os nomes das enzimas, foi decidido atribuir nomes relativos aos substratos que catalisam e contendo a terminação "-ase". Por exemplo, a amilase catalisa a hidrólise do amido e as proteases quebram ligações peptídicas.
Embora esta terminologia tenha simplificado a nomenclatura enzimática, a quantidade de enzimas conhecida (vários milhares) levou à criação de um sistema de divisão de diferentes tipos de enzimas. As enzimas dividem-se então em seis classes principais, de acordo com o tipo de reação química que catalisam:
| Classe 1 | Oxidorredutases | Catalisam reações de oxirredução, transferindo elétrons, hidretos (H-) ou prótons (H+). |
| Classe 2 | Transferases | Transferem grupos químicos entre moléculas. |
| Classe 3 | Hidrolases | Utilizam a água como receptor de grupos funcionais de outras moléculas. |
| Classe 4 | Liases | Formam ou destroem ligações duplas, respectivamente retirando ou adicionando grupos funcionais. |
| Classe 5 | Isomerases | Transformam uma molécula num seu isômero. |
| Classe 6 | Ligases | Formam ligações químicas por reações de condensação, consumindo energia sob a forma de ATP. |
Cada classe divide-se, por sua vez, em subclasses, também numeradas. As subclasses definem em que tipo de grupo as enzimas atuam. Por exemplo, um determinado grupo de enzimas pode pertencer à subclasse EC 2.1, que engloba "enzimas que transferem grupos contendo um carbono". As subclasses dividem-se ainda em sub-subclasses; continuando com o mesmo exemplo, existe a classe EC 2.1.1, que engloba as metiltransferases, ou seja, enzimas que transferem um grupo metilo. Finalmente, cada enzima recebe um quarto dígito específico à reação que catalisa; por exemplo, a enzima histamina N-metiltransferase tem o número EC 2.1.1.8 e catalisa especificamente a transferência de um grupo metilo para a histamina.
A nomenclatura das enzimas foi definida por uma comissão especializada, a Enzyme Committee (EC), que pertence à União Internacional de Bioquímica e Biologia Molecular (IUBMB). Cada enzima recebe então uma nomenclatura no formato EC X.Y.W.Z por esta razão. A Comissão de Nomenclatura da IUBMB (Nomenclature Committee of the International Union of Biochemistry and Molecular Biology, NC-IUBMB) é a responsável atual, em geral, pela nomenclatura de moléculas e processos relacionados à Bioquímica (e à Biologia Molecular).
Além do número EC, cada enzima possui um nome sistemático próprio, constituído pelos nomes dos substratos e da classe em que atuam. Usando o exemplo anterior da histamina N-metiltransferase (nome comum), esta enzima tem como nome sistemático S-adenosil-L-metionina:histamina N-tele-metiltransferase, indicando que a S-adenosil-L-metionina é o grupo dador, a histamina o aceitador, o grupo transferido é o metilo e a enzima é uma transferase. Os nomes comuns de enzimas são empregues de forma mais frequente que os sistemáticos para simplificação da escrita, em especial quando não existe possibilidade de confusão com outras enzimas (não existem, por exemplo, outras metiltransferases de histamina).
Estado de transição e equilíbrio químico
editarQualquer reação química pode ser descrita em termos energéticos: os reagentes possuem um determinado nível de energia, os produtos outro e, para que haja conversão de uma espécie química na outra, é necessário passar uma barreira energética. A barreira energética corresponde à existência do chamado estado de transição. Esta descrição pode ser feita graficamente:

em que ΔG é a variação da energia de Gibbs (ou da energia livre). Em Bioquímica, define-se a variação da energia livre padrão bioquímica, ΔG'º, uma forma ligeiramente diferente da variação da energia livre padrão, ΔGº: ambas são medidas à temperatura de 298 K (25ºC) e pressão de uma atmosfera mas, enquanto ΔGº considera a concentração de cada espécie química a 1M, a definição bioquímica simplifica para a condição em que o pH é igual a 7,0.
O equilíbrio que existe entre S e P depende da diferença entre os seus estados fundamentais de energia e é igual a ΔG'º. Se a energia do estado fundamental de P for menor que a de S, o equilíbrio S P é favorável à formação de P. Por muito elevada que seja essa diferença, S não é convertido a P a uma velocidade apreciável, ou sequer detectável, se a energia do estado de transição for muito elevada.
Porque existe esta barreira energética? Para que haja reação química, as moléculas do substrato têm de encontrar uma conformação tal que seja favorecida a reação. É necessário rearranjar geometrias, quebrar e formar ligações químicas e formar espécies intermediárias que, por não serem estáveis, possuem uma maior energia associada.
O estado de transição não corresponde à existência de um determinado intermediário reacional, sendo sim o ponto da reação em que esta pode progredir tanto na direção da formação de S como na de P. Desta forma, entre este ponto e o nível de energia dos substratos (ou dos produtos) existe uma diferença energética denominada energia de ativação, ΔG‡. Quanto mais elevada é esta energia, maior será a dificuldade em haver reação química.
As enzimas funcionam baixando precisamente esta energia de ativação, acelerando a reação. É de realçar mais uma vez que o equilíbrio da reação não é afetado; a enzima ajuda a reação a atingir mais rapidamente o equilíbrio mas não favorece nenhum dos sentidos da reação em particular.
Uma determinada reação química pode seguir de forma simples, havendo apenas um intermediário reacional, ou de forma mais complexa, em que existem dois ou mais intermediários reacionais. Neste contexto, um intermediário reacional é uma espécie química de curta existência, normalmente na ordem dos micro a milissegundos. A formação destas espécies pode ter diferentes energias de ativação; o passo da reação com maior energia de ativação prosseguirá com uma velocidade menor. Este facto condiciona a velocidade geral da reação e é por isso denominado passo limitante da reação. Na prática, uma reação que envolva vários intermediários não tem, normalmente, um único passo que seja exclusivamente limitante; os diferentes passos terão diferentes impactos na velocidade global da reação conforme a diferença entre as suas energias de ativação.
|
Esta página é um esboço de química. Ampliando-a você ajudará a melhorar o Wikilivros. |
Cinética enzimática
editarO estudo da função de enzimas envolve uma aproximação multidisciplinar. Por um lado, a determinação da estrutura, usando técnicas espectroscópicas como a difração de raios-X em proteínas cristalizadas ou a ressonância magnética nuclear (RMN) em proteínas em solução, permite localizar a posição dos diferentes resíduos no espaço. Este tipo de estudo revela muitas vezes detalhes sobre o mecanismo da reação, ou seja, a forma e sequência de ligações do substrato ao centro ativo para ser catalisado. Por outro lado, é importante conhecer parâmetros cinéticos, como a velocidade da reação catalisada, o efeito de inibidores nessa velocidade, quantas moléculas a enzima consegue catalisar por segundo, etc, para poder ser aferida a eficiência dessa enzima e a forma como é regulada.
Esta última aproximação ao estudo de enzimas, a cinética enzimática, é um dos campos de estudo mais clássicos da Bioquímica. Nenhum estudo sobre uma enzima se encontra completo sem estudos cinéticos.
Progressão da reação
editarPara que uma reação enzimática se dê, a enzima (E) e o substrato (S) têm de se encontrar e formar o complexo enzima-substrato (ES). Como referido anteriormente, embora este possa parecer um formalismo, a formação deste complexo é uma peça chave na compreensão da progressão de uma catálise. O complexo ES é lábil: a enzima pode dissociar-se do seu substrato sem ter havido reação. Este equilíbrio é expresso sob a forma:
- E+S ES
A enzima pode então transformar o substrato num produto (P). Existe de forma transiente um complexo EP, havendo então dissociação deste complexo em enzima livre (E) e produto (P). Este mecanismo pode escrever-se na forma simplificada:
- E+S [ES] E+P
A reação de formação de ES é normalmente mais rápida que a reação de dissociação de EP, ou seja, a libertação do produto da reação é normalmente um passo limitante da reação.
Velocidade inicial
editarO estudo de uma reação enzimática in vitro não reflete a verdadeira situação das enzimas em células, mas possibilita a simplificação da determinação de parâmetros cinéticos. Dentro de uma célula, o substrato pode estar confinado a um dado compartimento e não sofrer facilmente difusão: por exemplo, pode ser o produto de uma reação anterior numa via metabólica, pelo que passa diretamente de uma enzima para a outra. Normalmente, os estudos in vitro usam uma concentração de substrato muito superior à concentração de enzima, para que a probabilidade de uma enzima encontrar uma molécula de substrato seja o mais elevada possível.

Outra vantagem do uso de uma concentração de substrato ([S]) maior que a concentração de enzima ([E]) é a possibilidade de monitorizar a variação da velocidade de reação com [E] sem que haja uma variação apreciável de [S]. Como S está a ser convertido em P, [S] decresce com o tempo, à medida que o produto se acumula em solução. Como a [S] influencia a formação do complexo ES (e portanto da formação de produto), vai influenciar também a velocidade da reação. No entanto, se for feita apenas a monitorização da velocidade inicial da reação (tipicamente, durante os primeiros 30 a 60 segundos), a variação de [S] não é significativa e pode ser tomada como constante.
Nestas condições, a velocidade medida (velocidade inicial, V0) depende apenas de [E]. Um gráfico de V0 em função de [S] apresenta uma forma característica de semi-hipérbole: a baixas concentrações de substrato, a variação de V0 com [S] é linear; à medida que se usam [S] mais elevadas, V0 sofre uma variação cada vez menor, tendendo para um valor limite. Este valor é denominado velocidade máxima, Vmax.
A que se deve este comportamento? A baixas concentrações de substrato, a maior parte da enzima em solução encontra-se na forma livre, E; usando concentrações suficientemente elevadas de S, toda a enzima se encontrará, num dado instante, em complexo com molécula(s) de substrato, isto é, na forma ES. Por maior que seja a concentração de substrato utilizada, as moléculas de enzima não têm capacidade de catalisar o substrato, por todos os centros ativos se encontrarem ocupados. Nestas condições, diz-se que a enzima se encontra saturada.
Equação de Michaelis-Menten
editarA semi-hipérbole correspondente ao comportamento da velocidade de uma enzima em função da concentração do substrato pode ser descrita matematicamente pela equação de Michaelis-Menten:
![{\displaystyle V_{0}={\frac {V_{max}[S]}{K_{M}+[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/173b2b9318089a47e512c74cccfb4919625786be)
em que [S], V0 e Vmax são as grandezas anteriormente definidas e KM a constante de Michaelis. Esta equação pode ser deduzida de forma intuitiva considerando a forma como se desenrola a catálise enzimática.
É assumida, nesta dedução, a existência de um estado estacionário, em que [ES] permanece constante ao longo do tempo. A teoria do estado estacionário, introduzida por G. E. Briggs e J. B. S. Haldane em 1925, considera que a velocidade a que o complexo ES se forma é aproximadamente igual à velocidade da sua dissociação. Existe um curto período de tempo, normalmente na ordem dos microssegundos, em que há uma acumulação de ES – estado pré-estacionário. O estudo da cinética enzimática envolvendo a teoria do estado estacionário é referida como cinética do estado estacionário.
Também é assumido que a concentração de produto é negligível no início da reação, logo é negligível a extensão da reação inversa no equilíbrio entre substrato e produto, S P, considerando-se apenas a reação direta na dedução da equação. Assim, a reação é descrita por
- E+S [ES] E+P
Como já referido, a formação de [ES] limita a velocidade da reação, medida como V0:
- V0=k2[ES]
Se [ES] fosse facilmente mensurável, o valor de k2 seria simples de determinar diretamente. No entanto, esse não é o caso, e o valor de [ES] tem de ser deduzido. Para tal, considere-se que a concentração do total de enzima presente na reação, [E]t, é o somatório da concentração de enzima em complexo com o substrato, [ES], e da concentração de enzima "livre" (não ligada a substrato), [E]l:
- [E]t = [E]l + [ES]
Tratamento matemático e gráfico da equação de Michaelis-Menten
editarEmbora o gráfico obtido diretamente da equação de Michaelis-Menten seja de interpretação relativamente simples, existem tratamentos matemáticos que simplificam a representação gráfica da equação e permitem a obtenção rápida de parâmetros cinéticos.
O tratamento mais conhecido e porventura mais utilizado é o de Lineweaver-Burk. A equação de Michaelis-Menten pode ser transformada numa equação da reta, do tipo y=ax+b:
![{\displaystyle V_{o}={\frac {V_{m}ax[S]}{K_{M}+[S]}}\Leftrightarrow {\frac {1}{V_{o}}}={\frac {K_{M}+[S]}{V_{m}ax[S]}}\Leftrightarrow {\frac {1}{V_{o}}}={\frac {K_{M}}{V_{m}ax[S]}}+{\frac {[S]}{V_{m}ax[S]}}\Leftrightarrow {\frac {1}{V_{o}}}={\frac {K_{M}}{V_{m}ax[S]}}+{\frac {1}{V_{m}ax}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/856926843ff88c0198d4fac3979ccf522b871fdb)

Esta última forma da equação de Michaelis-Menten é conhecida como equação de Lineweaver-Burk. Um gráfico de (y) em função de (x) é uma reta de declive igual a (a), que intercepta o eixo das coordenadas na origem no ponto (b), interceptando também o eixo das abcissas no ponto Este gráfico é também chamado duplo recíproco pois trata-se de uma representação gráfica do recíproco de ambos os parâmetros Vo e [S].

![{\displaystyle {\frac {1}{[S]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/13eaed9b93d8ed8ac729f8dbb895987dbcc6597d)



Este tipo de tratamento matemático permite uma determinação mais precisa de Vmax, um parâmetro que só se obtém por aproximação num gráfico de Michaelis-Menten.
Outra vantagem na utilização da equação de Lineweaver-Burk é visível em estudos de inibição enzimática, sendo relativamente simples de detectar e distinguir entre diferentes tipos de inibição: ao usar diferentes concentrações de inibidores, e consoante o tipo de inibição efetuada, o gráfico de Lineweaver-Burk muda de uma forma bem estudada, como será mais claro no capítulo sobre inibição enzimática.
Enzimas alostéricas
Algumas enzimas não obedecem a cinética de Michaelis e Mentem, esse é o caso das enzimas que possuem uma espécie de modulador que no caso pode se apresentar com sinal positivo (a enzima está mais ativada)ou negativo (a enzima está menos ativada). São determinadas como passo limitante na maioria das vias-metabólicas exibindo assim como já dito anteriormente, a atividade mais ativada ou menos ativada em resposta a certos sinais.
A alosteria é explicada pelo efeito que o modulador + ou - age sobre a enzima, alterando assim a velocidade realizada pela enzima, possuem em geral uma grande relação com alguns distúrbios enzimáticos.
Existe a chamada inibição por retroalimentção ou Feedback, cujo há a existência de dois tipos:
-Homotrópico, na qual o sítio ativo da enzima pode funcionar como um regulador e o substrato como um modulador +.
-Heterotrópico, na qual o modulador é um metabólito diferente do substrato que podem se ligar a sítios distintos do substrato.
O modulador de forma resumida promove a facilitação da formação do complexo enzima-substrato, sofrendo algumas modificações covalentes reversíveis.
Glícidos
editarOs glícidos, também denominados hidratos de carbono, carbo-hidratos ou carboidratos, glúcidos, sacarídeos ou glicídios, são moléculas contendo vários grupos químicos funcionais hidroxilo e um aldeído ou cetona, ou polímeros hidrolisáveis constituídos por tais moléculas. São o grupo de moléculas existentes em sistemas vivos mais abundantes na Terra.
Os glícidos têm diversas funções, sendo as mais relevantes as funções estruturais e de armazenamento energético, especialmente na forma de polissacarídeos. São sintetizados através do processo fotossintético, entrando na composição de seres não fotossintéticos pela cadeia alimentar. Constituem a fonte primária de energia dos seres vivos.
O glícido mais comum é a glicose, que desempenha um papel fundamental na respiração celular e na fotossíntese.
Nomenclatura e composição química
editarQuimicamente, os glícidos são definidos como poli-hidroxi-aldeídos ou poli-hidroxi-cetonas. A designação "hidratos de carbono", hoje caída em desuso, provém do facto de a estrutura base dos glícidos ser uma cadeia de carbonos aos quais se encontram ligados átomos de oxigênio e hidrogênio na mesma proporção existente na molécula de água (ou seja, dois átomos de hidrogênio e um de oxigênio para cada átomo de carbono). Esta proporção é descrita pela fórmula empírica (CH2O)n. Nalguns casos, os glícidos podem conter nitrogênio ou enxofre.
Classificação
editarMonossacarídeos
editar
Os monossacarídeos têm fórmula estrutural (CH2O)n, em que "n" pode variar de 3 a 7:
- Triose: C3H6O3
- Tetrose: C4H8O4
- Pentose: C5H10O5
- Hexoses: C6H12O6
- Heptoses: C7H14O7,
sendo os mais importantes as pentoses e hexoses. Alguns dos monossacarídeos mais relevantes fisiologicamente incluem a glicose, a frutose, a galactose e a manose.
Para os seres vivos, as pentoses mais importantes são a ribose e a desoxirribose, que entram na composição química dos ácidos nucleícos, os quais comandam e coordenam as funções celulares.
As hexoses são monossacarídeos de 6 carbonos, que obedecem à fórmula geral C6H12O6. As hexoses mais importantes são a glicose, a frutose e a galactose, principais fontes de energia para os seres vivos. Ricas em energia, as hexoses constituem os principais combustíveis das células. São naturalmente sintetizadas na fotossíntese, processo de absorção de energia da luz; a reação geral é:
- 6 CO2 + 6 H2O -> C6H12O6 + 6O2
Oligossacarídeos
editarOligossacarídeos são pequenos polímeros constituídos por um reduzido número de monossacarídeos, tipicamente dois (dissacarídeos) ou três (trissacarídeos), normalmente não mais de dez. Oligossacarídeos mais longos estão geralmente associados a proteínas (glicoproteínas).
Exemplos de dissacarídeos incluem a sacarose e a lactose. A rafinose é um trissacarídeo comum.
A sacarose, o "açúcar de cana" ou de beterraba, é constituído por uma molécula de glicose ligada a uma frutose. A maltose é um dissacarídeo, pois é formada por duas moléculas de glicose. A lactose é encontrada somente no leite. Resulta da união de uma glicose com uma galactose.
Polissacarídeos
editar
Os polissacarídeos são cadeias longas, lineares ou ramificadas, de monossacarídeos. Os polissacarídeos mais importantes são o amido, a celulose e o glicogênio. O amido e o glicogénio actuam como reservas energéticas, enquanto que a celulose é um glícido estrutural. Outro polissacarídeo estrutural é a quitina, um componente fundamental do exoesqueleto de insetos.
Ao contrário dos mono e dos dissacarídeos, os polissacarídeos são insolúveis em água.
Monossacarídeos
editar
Os monossacarídeos, ou monossacáridos, são os glícidos mais simples. São constituídos por cadeias de carbono hidroxiladas, com a presença de grupos carbonila. A posição do grupo carbonila na cadeia permite distinguir duas famílias de monossacarídeos: as aldoses e as cetoses. As aldoses possuem o grupo carbonila na extremidade da cadeia de carbono (sendo então um grupo aldeído) e as cetoses possuem o grupo carbonilo num outro carbono, que não numa extremidade (sendo então um grupo cetona).
Fisicamente, são substâncias cristalinas, incolores, geralmente de sabor adocicado. São solúveis em água e insolúveis em solventes apolares. A assimetria de distribuição de grupos carbonila e hidroxila ao longo da cadeia leva à existência de diversos centros quirais, pelo que os monossacarídeos apresentam uma estereoquímica complexa.
Os monossacarídeos mais comuns possuem de três a sete carbonos na sua cadeia principal. Em solução aquosa, os monossacarídeos com cinco ou seis átomos de carbono tendem a formar estruturas cíclicas.
Nomenclatura
editarOs monossacarídeos podem ser genericamente designados pela família a que pertencem e quanto ao número de carbonos da sua cadeia. Assim, monossacarídeos com três carbonos são trioses, com quatro carbonos tetroses, tendo-se pentoses, hexoses e heptoses se possuírem cinco, seis ou sete átomos de carbono, respectivamente. É acrescentado o prefixo "aldo" ou o prefixo "ceto" conforme a sua família das aldoses ou das cetoses, respectivamente. Pode ter-se então, por exemplo, aldotrioses, aldopentoses, ceto-hexoses, etc. Note-se a presença do sufixo "-ose", comum aos oligossacarídeos (que por vezes são também designados simplesmente "oses").
Alguns dos monossacarídeos mais representativos encontram-se na figura abaixo. Dentre estes, destaque-se a D-glucose (uma aldo-hexose), a D-frutose (uma ceto-hexose) e o D-gliceraldeído (uma aldotriose).
-
 D-manitol (aldo-hexose)
D-manitol (aldo-hexose) -
 D-xilose (aldopentose)
D-xilose (aldopentose) -
 L-frutose (ceto-hexose)
L-frutose (ceto-hexose) -
 D-gliceraldeído (aldotriose)
D-gliceraldeído (aldotriose) -
 D-ribulose (cetopentose)
D-ribulose (cetopentose) -
 D-fucose (aldo-hexose)
D-fucose (aldo-hexose)






Quiralidade e fórmula de Fischer
editarExceto a di-hidroxiacetona, todos os monossacarídeos possuem átomos de carbono quirais, ou seja, em que os quatros grupos diretamente ligados ao carbono são diferentes, podendo então apresentar diferentes conformações. O gliceraldeído possui apenas um centro quiral, apresentando dois possíveis enantiômeros, designados D-gliceraldeído e L-gliceraldeído. Ao desenhar a fórmula estrutural do gliceraldeído no papel, é possível fazê-lo de duas formas fundamentalmente diferentes: colocando o grupo aldeído no topo e o carbono não-quiral no extremo oposto, o grupo hidroxilo pode ser colocado do lado direito (isômero D) ou do lado esquerdo (isômero L).
-
 Projeção de Fischer do D-gliceraldeído.
Projeção de Fischer do D-gliceraldeído. -
 Projeção de Fischer do L-gliceraldeído.
Projeção de Fischer do L-gliceraldeído.


Esta forma de representação da fórmula estrutural é conhecida como fórmula de projeção de Fischer, ou mais simplesmente fórmula de Fischer. A disposição dos átomos no plano não é aleatória: assume-se que as ligações químicas desenhadas na vertical estão atrás do plano do desenho, enquanto que as ligações químicas desenhadas horizontalmente estão projetadas acima desse plano. As moléculas são representadas com o grupo carbonilo o mais próximo possível do topo.
A fórmula de Fischer permite comparar de forma simples estruturas de monossacarídeos entre si e com o gliceraldeído. Considerando-se o centro quiral mais distante do grupo carbonilo, compara-se a posição do grupo -OH nesse carbono com a posição do grupo -OH nos diferentes enantiômeros de gliceraldeído: atribui-se então a nomenclatura D- e L- conforme a semelhança com o respectivo enantiômero do gliceraldeído. Uma molécula com n centros quirais terá 2n estereoisômeros; por exemplo, existem dezasseis aldo-hexoses, sendo oito enantiômeros L- e oito D-. No entanto, a maioria das aldo-hexoses encontradas em sistemas vivos são D-hexoses.
Os carbonos da cadeia dos monossacarídeos são numerados seguindo a nomenclatura clássica da Química Orgânica, começando-se no carbono mais próximo do grupo de maior prioridade. Neste caso, tal grupo é o carbonilo. Cada estereoisômero tem, no entanto, um nome trivial: têm-se a D-glucose, a D-manose, a D-galactose como as aldo-hexoses mais importantes, existindo outras como a D-talose ou a D-gulose, por exemplo. As tetracetoses e as pentacetoses obtêm o seu nome a partir das tetra-aldoses e penta-aldoses correspondentes, inserindo a partícula "ul" no meio no nome: por exemplo, a D-ribulose é a cetose correspondente à aldose D-ribose e a D-xilulose é a cetose correspondente à aldose D-xilose.
Alguns monossacarídeos são muito semelhantes, diferindo apenas na conformação quiral de um carbono; tais pares de monossacarídeos são chamados epímeros. A D-glucose e a D-galactose são um exemplo.
Estrutura cíclica
editarA representação estrutural de Fischer é útil para a distinção de enantiômeros, mas na realidade os monossacarídeos adotam uma conformação cíclica quando em solução aquosa. Tal conformação ocorre através da ligação covalente do grupo carbonilo com um dos grupos hidroxilo da própria molécula. Esta reação ocorre com as aldotetroses e com todos os monossacarídeos com cinco ou mais átomos de carbono.
A reação de um grupo carbonilo com um grupo hidroxilo resulta na formação de compostos conhecidos como hemiacetais (se o grupo carbonilo é um aldeído) ou hemicetais (se o grupo carbonilo é uma cetona). As aldo-hexoses podem formar através desta reação anéis de cinco ou seis membros, conforme o grupo hidroxilo com que o aldeído reage. Pela sua semelhança aos compostos orgânicos furano e pirano, as aldo-hexoses que formam compostos cíclicos são denominadas furanoses e piranoses, com estrutura em anel de cinco e seis membros, respectivamente. As piranoses são, no entanto, mais estáveis em solução e constituem a forma predominante em organismos vivos.
Após a reação que forma o hemiacetal ou o hemicetal, podem existir diferentes posições do grupo -OH formado nesta reação pela redução do carbonilo. Estes diferentes isômeros são denominados anômeros, podendo interconverter-se em solução numa reação chamada de mutarrotação.
Fórmulas de Haworth e fórmulas conformacionais
editarPara maior clareza sobre a estrutura e, principalmente, sobre a estereoquímica das furanoses e piranoses, é comum usar-se a fórmula de perspectiva de Haworth. Nesta, o anel é colocado de forma semelhante à do pirano ou do furano, mas com um dos lados traçado de forma mais espessa, para demonstrar perspectiva (esse lado estará mais próximo do leitor). Visualiza-se então um plano formado pelo anel, acima e abaixo do qual se projetam os restantes grupos; os que estão acima do anel estariam no lado esquerdo numa projeção de Fischer.

No entanto, o anel não é planar, como pode ser sugerido pela fórmula de perspectiva de Haworth. Tais anéis tendem a tomar uma de duas conformações, devido à repulsão das nuvens eletrônicas dos átomos dos grupos ligados à cadeia principal: conformação em cadeira e conformação em barco, sendo a conformação em cadeira a de menor energia (menor repulsão entre as nuvens eletrônicas).

Existe conversão entre as duas conformações em solução aquosa.
Oligossacarídeos
editarOs oligossacarídeos são constituídos por um reduzido número de monossacarídeos, tipicamente dois (dissacarídeos) ou três (trissacarídeos). Os dissacarídeos são o grupo mais representativo desta classe.
Os oligossacarídeos são primariamente uma fonte de energia para os organismos vivos. Existem oligossacarídeos que participam em funções estruturais, ao serem ligados a proteínas (glicoproteínas).
Nomenclatura
editarAlguns oligossacarídeos de maior importância são os dissacarídeos sacarose, maltose e lactose. Estes são nomes triviais; a nomenclatura oficial segue as seguintes regras:
- determina-se a conformação do carbono anomérico que efetua a ligação (se é α ou β);
- determina-se o nome do resíduo não redutor, inserindo a partícula "furano" ou "pirano" conforme a forma do anel. Por exemplo, fructofuranose;
- indica-se os números dos dois carbonos que são ligados, entre parênteses, ligados por uma seta. Por exemplo, (1→2) indica uma ligação entre o carbono C1 de uma molécula e o C2 da segunda;
- nomeia-se então o segundo resíduo.
A regra repete-se quando os oligossacarídeos são constituídos por mais de duas unidades. Os nomes sistemáticos podem tornar-se então muito longos, sendo por vezes preferível a adoção de abreviaturas. Como a maioria dos enantiômeros encontrados em sistemas vivos são D-açúcares e os mais interessantes as piranoses, em geral obvia-se a menção destes dois parâmetros. Por exemplo a lactose passa a ser Gal(β1→4)Glc em vez de β-D-galactopiranosil-(1→4)-β-D-glicopiranose.
Dissacarídeos
editarUm dissacarídeo é formado por dois monossacarídeos ligados através de uma reação de condensação. No processo, é libertada uma molécula de água. A ligação assim obtida é denominada O-glicosídica, correspondendo à formação de um acetal a partir de um hemiacetal e de um grupo álcool (hidroxilo) de uma segunda molécula de açúcar. A reação reversa (de separação do dissacarídeo em dois monómeros) é a hidrólise. Os dissacarídeos têm a fórmula geral C12H22O11.
A sacarose, o vulgar açúcar usado em culinária, é constituído por uma molécula de glicose ligada a uma de frutose. A maltose é também um dissacarídeo, formada por duas moléculas de glicose. A lactose é encontrada no leite e resulta da união de uma glicose com uma galactose.
Sacarose
editar
A sacarose é composta por uma molécula de α-D-glicose e uma de β-D-frutose, tendo os átomos de carbono C1 da glicose e C2 da frutose participando na ligação glicosídica. O seu nome sistemático é α-D-glucopiranosil-(1→2)-β-D-fructofuranose (abreviado Glc(α1→2β)Fru). É conhecida como o comum açúcar, sendo extraído para distribuição comercial da cana-do-açúcar (Sacharum officinarum) e da beterraba (Beta vulgaris).
A sacarose é um açúcar não redutor pois ambos os grupos químicos de natureza redutora dos monômeros que a constitui participam na ligação glicosídica. Assim, a sacarose não pode formar polímeros. A ligação glicosídica pode ser hidrolisada, mas é muito estável: uma solução aquosa de sacarose mantém-se estável durante vários anos. Um conjunto de diferentes enzimas conhecidas como sacarases hidrolisam a ligação glicosídica da sacarose a uma velocidade muito superior.
Em mamíferos, a sacarose é digerida no estômago por hidrólise ácida. A hidrólise ácida é também usada em laboratório para obter glucose e frutose da sacarose. A sacarose que não é digerida até chegar ao intestino delgado pode ser hidrolisada por outras glicosilases presentes nas membranas citoplasmáticas das células epiteliais de microvilosidades no duodeno. A glucose e frutose resultantes da hidrólise da sacarose são rapidamente absorvidas no intestino delgado para a corrente sanguínea. É por isso uma fonte de energia química rápida (com um conteúdo de 17kJ/g), causando a subida nos níveis sanguíneos de glucose logo após a ingestão.
Em bactérias e alguns animais, a enzima invertase hidrolisa a sacarose.
Maltose
editar
A maltose (nome sistemático α-D-glicopiranosil-(1→4)-D-glicopiranose, Glc(α1→4)Glc) é formada por duas moléculas de glicose ligadas por uma ligação glicosídica α(1→4). Pode polimerizar-se com mais monômeros de glucose, formando pequenos polímeros conhecidos como dextrinas (ou maltodextrinas) e eventualmente amido.
Tal como a sacarose, a maltose pode ser hidrolisada com ácido ou enzimaticamente, neste caso pela enzima maltase, resultando duas moléculas de glicose por molécula de maltose.
A maltose é produzida em cereais em germinação, tais como a cevada, num processo relevante na fermentação alcoólica de bebidas. Na maltagem da cevada, a concentração de malte é maximizada neste cereal; a maltose é então usada pelas leveduras durante a fermentação, produzindo etanol e dióxido de carbono.
Lactose
editar
A lactose (β-D-galactopiranosil-(1→4)-β-D-glicopiranose, Gal(β1→4)Glc) é composta por uma molécula de β-D-glicose e uma de β-D-galactose ligadas por uma ligação glicosídica β(1→4). É conhecida vulgarmente por "açúcar do leite" pois constitui entre 2 e 8% dos sólidos presentes no leite.
A hidrólise da lactose é feita pela enzima lactase (β(1→4) dissacaridase), produzida nas vilosidades intestinais. A glucose e galactose resultantes são absorvidas para a corrente sanguínea.
Como a principal ocorrência da lactose é no leite, na maioria dos mamíferos a produção de lactase decresce gradualmente com a idade, passando a ser incapazes de metabolizar a lactose. Alguns povos têm porém uma versão de lactase que é funcional após a infância; o leite faz parte da dieta humana em diversas regiões do globo.
Trealose
editar
A trealose (α-D-glicopiranosil-α-D-glicopiranose, Glc(α1→1α)Glc) é um dissacarídeo não redutor de glicose, tal como a sacarose. Está presente na hemolinfa (sistema circulatório) de insetos, sendo uma fonte de energia rapidamente disponível para o voo. Pode também ser sintetizada por fungos e plantas. Pode ser hidrolisada pela enzima trealase, originando duas moléculas de glicose por uma de trealose.
Polissacarídeos
editarOs polissacarídeos são polímeros longos constituídos por monossacarídeos. Enquanto que mono e oligossacarídeos são usados no metabolismo energético, os polissacarídeos estão envolvidos não só no metabolismo energético como também na construção de estruturas celulares. Os açúcares simples têm uma disponibilidade imediata para suprir déficits energéticos num organismo; em contraste, os polissacarídeos são reservas de energia, em que a sua hidrólise é necessária para haver disponibilidade de açúcares no metabolismo.
Quanto à sua constituição química, os polissacarídeos podem ser divididos em homopolissacarídeos (constituídos por um só tipo de monômero) e heteropolissacarídeos (constituídos por diferentes tipos de monômeros).
Celulose
editar
A celulose, conhecida vulgarmente por fibra, encontra-se apenas em plantas, sendo um constituinte das paredes celulares. É um homopolissacarídeo de β-glicose, o que lhe confere uma estrutura mais rígida que ligações entre α-glicose. Os mamíferos são incapazes de digerir a celulose mas esta é uma importante parte da dieta humana, ao ajudar à limpeza do tracto intestinal.
Amido
editar

O amido é a reserva energética das plantas. É um homopolissacarídeo formado por α-glicose. Encontra-se em duas formas: amilose e amilopectina.
Glicogênio
editarO glicogênio é um homopolissacarídeo ramificado, constituído por glicose, sendo a principal reserva energética em animais. Os humanos têm a capacidade de armazenar pequenas quantidades de glicogênio no fígado e nos músculos. É sintetizado quando os níveis de glicose no sangue são altos; essa síntese é estimulada pela insulina, produzida no pâncreas.
|
Esta página é um esboço de química. Ampliando-a você ajudará a melhorar o Wikilivros. |
Lípidos
editar
Os lipídios (ou lípidos) são moléculas orgânicas anfipáticas (há exceções) contendo cadeias de hidrocarbonetos, que são responsáveis pela sua baixa solubilidade em água. A palavra vem do grego lipos, que significa gordura.
Os lipídios têm várias funções biológicas diversas como a formação de membranas, reserva energética (devido à forma altamente reduzida do carbono (hidrocarboneto) e sinalização celular.
Índice
editarNeste capítulo será abordado diferentes classes desta molécula e suas respectivas funções.
Ácidos gordos
editar


Os ácidos gordos (Portugal) ou ácidos graxos (Brasil) podem ser caracterizados quimicamente por conterem um ácido carboxílico ligado a uma "cauda" de hidrocarboneto que varia em tamanho e saturação. Na natureza, os ácidos palmítico e esteárico (16 e 18 carbonos respectivamente) são os ácidos gordos saturados mais abundantes. Do ponto de vista energético, o ácido gordo é um excelente "reservatório" de energia, haja visto que os carbonos que o compõe estão extremamente reduzidos.
Os ácidos gordos cumprem diversas funções: são precursores de outros lípidos, como os eicosanóides, e fazem parte da constituição de lípidos estruturais, como os fosfolípidos. Além disso, a sua oxidação a CO2 e H2O liberta uma grande quantidade de energia metabólica.
Possuem normalmente entre quatro e 36 carbonos na cadeia alifática. Esta pode conter apenas ligações simples ou apresentar ligações duplas em alguns pontos; também pode apresentar pequenas ramificações (grupos metilo), anéis com três carbonos e grupos hidroxila.
Tipos de ácidos gordos
editarÁcidos gordos saturados
editarOs ácidos gordos saturados são aqueles que possuem ligação simples entre seus átomos de carbono ou outro grupo funcional ao longo da cadeia. Geralmente possuem uma forma reta, o que permite seu armazenamento de forma muito eficiente.
Veja alguns do ácidos gordos mais comuns na tabela abaixo:
| Nome comum | Nome sistemático | Estrutura | Notação | Ponto de fusão (°C) |
| Ácido acético | Ácido etanóico | CH3COOH | 2:0 | 16,5 |
| Ácido propiônico | Ácido propanóico | CH3(CH2)COOH | 3:0 | |
| Ácido butírico | Ácido butanóico | CH3(CH2)2COOH | 4:0 | |
| Ácido valérico | Ácido propilacético | CH3(CH2)3COOH | 5:0 | |
| Ácido capróico | Ácido hexanóico | CH3(CH2)4COOH | 6:0 | |
| Ácido caprílico | Ácido octanóico | CH3(CH2)6COOH | 8:0 | |
| Ácido cáprico | Ácido decanóico | CH3(CH2)8COOH | 10:0 | |
| Ácido láurico | Ácido dodecanóico | CH3(CH2)10COOH | 12:0 | 44,2 |
| Ácido mirístico | Ácido tetradecanóico | CH3(CH2)12COOH | 14:0 | 53,9 |
| Ácido palmítico | Ácido hexadecanóico | CH3(CH2)14COOH | 16:0 | 63,1 |
| Ácido esteárico | Ácido octadecanóico | CH3(CH2)16COOH | 18:0 | 69,6 |
| Ácido araquídico | Ácido eicosanóico | CH3(CH2)18COOH | 20:0 | 76,5 |
| Ácido beênico | Ácido docosanóico | CH3(CH2)20COOH | 22:0 | 86,0 |
| Ácido lignocérico | Ácido tetracosanóico | C24H48O2 | 24:0 | 84,0 |
| Ácido palmitoleico* | Ácido cis-9-hexadecenóico | CH3(CH2)5CH=CH(CH2)7COOH | 16:1Δ9 | 1 - 0,5 |
| Ácido oleico* | Ácido cis-9-octadecenóico | CH3(CH2)7CH=CH(CH2)7COOH | 18:1Δ9 | 13,4 |
| Ácido linoleico* | Ácido cis-,cis-9,12-octadecadienóico | CH3(CH2)4CH=CHCH2CH=CH(CH2)7COOH | 18:2Δ9,12 | 1 - 5 |
*Ácidos gordos insaturados
O termo "saturado" se refere ao hidrogênio, isto é, um ácido gordo saturado é aquele no qual todos os carbonos (da cadeia de hidrocarbonetos) estão ligados a dois hidrogênios, exceto ao último carbono que deve estar ligado a três hidrogênios.
Ácidos gordos insaturados
editarOs ácidos gordos insaturados seguem o mesmo padrão dos ácidos gordos saturados, exceto pela existência de uma ou mais duplas ligações ao longo da cadeia. A dupla ligação ocorre entre carbonos (-CH=CH-) e de forma alternada, isto é, um único átomo de carbono só forma uma dupla ligação (do tipo -CH=CH-CH=CH- e nunca -CH=C=CH).
A dupla ligação pode ter duas configurações; se o ácido graxo adquirir uma forma "linear", é dito que a ligação tem uma "configuração trans", mas se o ácido gordo forma uma "quina" a ligação possui configuração cis.
Uma configuração cis quer dizer que os átomos de carbonos adjacentes estão do mesmo lado da dupla ligação. A rigidez da dupla ligação torna o ácido gordo menos flexível. Quanto maior for o número de duplas ligações, maior é a curva do ácido gordo. Um notável papel desempenhado pela ligação cis ocorre nas membranas biológicas: como essas membranas são constituídas por lipídios e esses, na sua maioria, possuem ácidos gordos como constituintes estruturais, o número total de ligações cis em uma membrana vai influenciar sua fluidez (flexibilidade).
Já uma configuração trans, por sua vez, significa que os dois átomos de carbonos em ambas as extremidades da dupla ligação estão do lado oposto. Como consequência, não há dobramento de cadeia, e sua conformação é muito semelhante a de um ácido gordo saturado.
Os ácidos gordos insaturados de ocorrência natural normalmente possuem configuração cis. A maioria dos ácidos gordos de configuração trans não é encontrada na natureza e sim em gorduras que passaram por processos artificiais, especialmente como produto minoritário da hidrogenação de gorduras insaturadas (que consiste em reduzir as ligações duplas de ácidos cis a ligações simples).
Alimentos contendo ácidos gordos insaturados podem sofrer, por exposição prolongada ao ar, oxidação nas ligações duplas, resultando na quebra da cadeia de carbonos na zona dessa ligação e consequente formação de aldeídos de cadeia curta, de sabor e odor desagradável (o ranço). tanto a cadeia de ácidos saturados, quanto a cadeia de ácidos insaturados são importantes para manterem a membrana em equilíbrio, assim desenvolvendo as suas funções.
Nomenclatura
editar
O carbono da cadeia hidrofóbica diretamente ligado ao carbono do grupo carboxílico é convencionado como carbono-alfa (α), enquanto que o carbono da extremidade oposta é o carbono-ômega (ω). A numeração dos carbonos inicia-se no carbono do grupo carboxílico; o carbono-α é então o carbono-2.
A presença de ligações duplas numa cadeia de ácido gordo é denotada pelo seu número separado do número de carbonos da cadeia principal por dois pontos. Por exemplo, o ácido oleico, que contém 18 carbonos e uma ligação dupla, denota-se 18:1. O ácido esteárico tem o mesmo número de carbonos, mas nenhuma ligação dupla, sendo por isso 18:0.
É também normalmente interessante denotar a posição das ligações duplas ao longo da cadeia. Assim, a posição é denotada por um delta (Δ) em sobrescrito. Por exemplo, um ácido gordo com uma cadeia de 22 carbonos e duas ligações duplas, uma entre os carbonos 9 e 10 e outra entre os carbonos 13 e 14, será denotado 22:2(Δ9,13).
Os ácidos gordos mais comuns têm número par de carbonos; as ligações duplas, se presentes, raramente são conjugadas e encontram-se normalmente entre os carbonos 9 e 10 e também entre os carbonos 12 e 13 e os carbonos 14 e 15. Os ácidos gordos insaturados produzidos naturalmente têm usualmente as suas ligações duplas na conformação cis; em determinadas condições (como nas reações de hidrogenação de alguns óleos ou no processo digestivo de ruminantes), são formadas ligações trans. As chamadas "gorduras trans" devem o seu nome a eta característica e são consideradas prejudiciais à saúde por o seu consumo estar ligado ao aumento de colesterol "mau" (LDL).
Os ácidos gordos apresentam baixa solubilidade em água e são mais insolúveis quanto maior for a cadeia alifática e menor o número de ligações duplas nesta.
Comportamento em solução
editar
De acordo com AMABIS (1994), Em solução aquosa, os ácidos gordos tendem a agrupar-se em estruturas globulares denominadas micelas, em que a zona do ácido carboxílico ("cabeça") se situa na zona externa da micela, por ser a parte hidrofílica da molécula, enquanto que as "caudas" (hidrofóbicas) apontam para o interior da micela, evitando o contato com o solvente polar. Também pode ocorrer a formação de bicamadas, com a mesma organização das micelas mas estruturando-se formando duas camadas, com as "caudas" voltadas para o lado interno da bicamada. A estrutura em bicamada permite a existência de membranas que compartimentalizam a célula e a separam do meio exterior.
A presença de ácidos insaturados na bicamada lipídica influencia a fluidez da membrana. Uma bicamada composta apenas por ácidos gordos saturados apresenta um empacotamento denso, em que as cadeias longas de carbonos interagem intimamente através de ligações de van der Waals. Por esta razão, gorduras saturadas de cadeia longa são normalmente sólidas à temperatura ambiente (25°C). A presença de ligações duplas cis (mas não trans) introduz um ângulo na estrutura do ácido gordo que diminui a ordem do empacotamento e o número de interações de van der Waals entre as cadeias. Como consequência, a estrutura da bicamada torna-se mais "aberta" e fluida, sendo necessária menos energia para perturbar o empacotamento dos ácidos gordos. Por esta razão, gorduras insaturadas são usualmente líquidas à temperatura ambiente. (AMABIS, 1994)

- Obs.: Esta página encontra-se em Português europeu. Alguns termos, como "ácido gordo", podem ser traduzidos, no Brasil, como "ácidos graxos".
Os acilgliceróis (ou gorduras apolares) contêm glicerol (propanotriol) unido a um ou vários ácidos gordos através de ligações éster. Os mais abundantes são os triglicéridos (triacilgliceróis), também simplesmente chamados gorduras (ou ainda gorduras neutras). Geralmente o ácido gordo insaturado, quando existente, localiza-se no meio da molécula de triglicérido. Existem também diacilgliceróis (diglicéridos), possuindo apenas dois ácidos gordos.

Os triacilgliceróis podem ser simples ou mistos, conforme sejam compostos por apenas um tipo de ácido gordo ou diferentes ácidos gordos. Os triacilgliceróis simples recebem nomes correspondentes aos ácidos gordos que os compõem. Por exemplo, a trimistirina é um triacilglicerol simples contendo três ésteres de moléculas de ácido mirístico. Quando os ácidos gordos ligados aos carbonos 1 e 3 da molécula de glicerol num triacilglicerol misto são diferentes, este apresenta quiralidade.

Propriedades físicas
editarApesar de o glicerol e os ácidos gordos serem moléculas polares, os triacilgliceróis não o são. Os grupos polares do glicerol e dos ácidos formam as ligações éster que os unem. Como resultado, os triacilgliceróis são muito insolúveis em água. Têm também uma densidade inferior à da água à temperatura ambiente.
No metabolismo e alimentação
editarOs organismos vertebrados armazenam estas gorduras em células especializadas chamadas adipócitos, que são quase totalmente preenchidas por gotículas de triacilgliceróis. Tanto em plantas como em animais, servem como uma eficiente reserva de energia metabólica.
Obtém-se mais energia metabólica a partir de uma determinada quantidade de triacilgliceróis do que da mesma quantidade de polissacarídeos essencialmente por duas razões:
- Os polissacarídeos, quando armazenados na célula, precisam de se encontrar hidratados, ou seja, têm diversas moléculas de água associadas (2 g de água por cada grama de polissacarídeo). Em contrapartida, os triacilgliceróis, por serem apolares, estão armazenados sem essa camada de hidratação, de forma mais condensada nos adipócitos.
- Por serem compostos maioritariamente por longas cadeias de carbonos ligadas apenas a hidrogênios, os triacilgliceróis encontram-se num estado mais reduzido que os polissacarídeos (que possuem diversos átomos de oxigênio). Assim, ao fazer-se a oxidação destes compostos, obtém-se mais do dobro da energia a partir de triacilgliceróis que de polissacarídeos, por grama de composto.
Apesar de ambos serem utilizados como reservas de energia, os polissacarídeos e as gorduras têm funções essencialmente diferentes. Quando o organismo precisa de energia rapidamente disponível, recorre às reservas de polissacarídeos: o seu catabolismo origina açúcares facilmente assimiláveis e utilizáveis em vias catabólicas como a glicólise. Por outro lado, as gorduras são reservas de "longo prazo", de catabolismo mais lento. Além desta função, as reservas de gorduras são essenciais no isolamento térmico de alguns animais, como o urso polar: é a camada adiposa que permite resistir às baixas temperaturas árticas, atuando como isolante térmico.
Encontram-se triacilgliceróis em diversos alimentos. A maior fonte na alimentação provém de óleos vegetais, como o azeite ou o óleo de sésamo (tipicamente contendo triacilgliceróis insaturados) e gorduras animais (tipicamente saturadas).
Fosfolípidos
editarUm dos principais componentes estruturais da membrana celular são os fosfolípidos. Os fosfolípidos mais comuns são os glicerofosfolípidos, compostos por dois ácidos gordos e um grupo fosfato ligados a uma molécula de glicerol por ligações éster, à semelhança do que acontece com os triacilgliceróis. Existe ainda um grupo polar adicional ligado ao glicerol através do fosfato: esta ligação ao glicerol é, mais especificamente, uma ligação fosfodiéster. Existem também os esfingolípidos, que utilizam esfingosina em vez de glicerol (e contêm apenas um ácido gordo). No entanto, nem todos os esfingolípidos possuem um grupo fosfato (ou seja, nem todos são considerados fosfolípidos).

A presença do grupo fosfato e do grupo polar a ele ligado nestas moléculas confere-lhes uma característica muito importante: em vez de serem totalmente apolares, como os triacilgliceróis, são anfipáticas, ou seja, possuem uma zona polar e uma apolar. Numa solução aquosa, moléculas anfipáticas tendem a agrupar-se de forma a que as suas partes apolares não estejam, tanto quanto possível, em contato direto com as moléculas de água. Os grupos polares, no entanto, "preferem" estar em contato com a água. Isto acontece por a água ser também uma molécula polar, como já se viu no capítulo sobre esta molécula. Por causa desta propriedade, os fosfolípidos tendem a agrupar-se de duas formas distintas:
- em bicamada, ou seja, formando uma "sanduíche" em que as "cabeças" polares dos fosfolípidos se encontram em contato com o meio externo (aquoso) e as "caudas" apolares se refugiam entre as duas camadas formadas pelas "cabeças";
- em micela, em que existe apenas uma camada de fosfolípidos; para que as caudas apolares não estejam em contato com o meio aquoso, estas estruturas tomam uma forma esférica.
Glicerofosfolípidos
editarComo referido acima, os glicerofosfolípidos são os fosfolípidos mais abundantes em membranas celulares. Diferentes grupos polares podem ligar-se ao glicerol através da ligação fosfodiéster, conferindo diferentes cargas elétricas à "cabeça" do fosfolípido e, consequentemente, à superfície da membrana celular. É também a presença deste grupo que determina a nomenclatura trivial dos glicerofosfolípidos. Por exemplo, se o grupo for colina, teremos fosfatidilcolina; se for serina, fosfatidilserina, etc. Existem na realidade diversas fosfatidilcolinas, fosfatidilserinas, etc, dependendo dos ácidos gordos que compõem a parte apolar da molécula, pelo que a diversidade de glicerofosfolípidos pode ser muito grande. Em geral, possuem um ácido gordo saturado ligado ao carbono 1 do glicerol e um insaturado no carbono 2 (estando o grupo fosfato ligado ao carbono 3).
Encontram-se na tabela abaixo alguns exemplos de glicerofosfolípidos:
| Nome | Grupo polar | Fórmula estrutural |
|---|---|---|
| Ácido fosfatídico | hidrogênio | 
|
| Fosfatidiletanolamina | etanolamina | 
|
| Fosfatidilcolina | colina | 
|
| Fosfatidil-L-serina | L-serina | 
|
| Fosfatidilglicerol | glicerol | 
|
| Fosfatidilglicerofosfato | fosfato de glicerol | |
| Cardiolipina (difosfatidilglicerol) |
fosfatidilglicerol | |
| Fosfatidilinositol-3,5-bisfosfato | 3,5-bisfosfato de inositol | 
|
Esfingolípidos contendo fosfato (esfingomielina)
editar
Os esfingolípidos são estruturalmente semelhantes aos glicerofosfolípidos, possuindo uma cabeça polar e duas caudas apolares. No entanto, em vez de glicerol, possuem esfingosina, e uma das caudas não é um ácido gordo mas sim parte da cadeia de esfingosina.

A esfingosina é um aminoálcool linear de cadeia longa (dezoito carbonos) que, como o nome indica, possui um grupo álcool (-OH) e um amina (-NH2) na sua estrutura, além de uma ligação dupla entre os carbonos 4 e 5. O grupo amina liga-se a um ácido gordo, enquanto que o grupo álcool se liga a um grupo polar.
Existem esfingolípidos contendo fosfato ligado a um grupo polar, tal como nos glicerofosfolípidos, e esfingolípidos sem fosfato. Nesta seção descrevem-se os primeiros, normalmente conhecidos como esfingomielinas; os restantes esfingolípidos serão abordados no capítulo sobre outros lípidos de membrana.
As esfingomielinas possuem, como dito, um grupo polar ligado à esfingosina através de uma ligação fosfodiéster. O grupo polar é normalmente colina ou etanolamina (o conjunto grupo polar + grupo fosfato é geralmente designado fosfocolina e fosfoetanolamina, respectivamente). Comportam-se estruturalmente como os glicerofosfolípidos, encontrando-se em membranas de células animais.

As ceras são lípidos que por hidrólise libertam 1 mol de ácido gordo de cadeia longa e 1 mol de álcool alifático de cadeia longa por mole de cera. Tal como os acilgliceróis, os ácidos gordos e os álcoois são ligados por ligações éster.
Possuem estrutura linear, o que facilita a agregação entre as moléculas, formando cadeias hidrofóbicas. Têm um alto ponto de fusão (entre 60 e 100ºC). Estas características conferem funções impermeabilizantes e estruturais às ceras.
São segregadas por diversos organismos, particularmente em situações que necessitem de resistência à evaporação de água. Por exemplo, as aves possuem glândulas que segregam ceras utilizadas para impermeabilização das suas penas. Também é conhecida a cera de abelha, utilizada por este insecto para construir os favos em colmeias, necessários para a sua reprodução.
|
Esta página é um esboço de química. Ampliando-a você ajudará a melhorar o Wikilivros. |
Esfingolípidos são lípidos complexos em que o glicerol é substituído pela esfingosina,que é um aminoácido de cadeia longa e hidroxilada. Normalmente atuam como antigênios de superfície nas biomembranas.
|
Esta página é um esboço de química. Ampliando-a você ajudará a melhorar o Wikilivros. |
O que são vitaminas
editarAs vitaminas são nutrientes importantes para o nosso organismo. São de extrema importância para o bom funcionamento do nosso organismo, principalmente, porque ajuda a evitar muitas doenças.
Elas não são produzidas pelo organismo e, portanto, devem ser adquiridas através da ingestão de alimentos (frutas, verduras, legumes, carnes etc). A falta de vitaminas pode acarretar em diversas doenças (avitaminoses). Elas podem ser de dois tipos: hidrossolúveis (solúveis em água e absorvidas pelo intestino) e lipossolúveis (solúveis em gorduras e absorvidas pelo intestino com a ajuda dos sais biliares produzidos pelo fígado).
Classificação das vitaminas
editarHidrossolúveis
- tiamina (vitamina B1)
- riboflavina (vitamina B2)
- ácido pantotênico (vitamina B5)
- piridoxina, piridoxamina e piridoxal (Vitamina B6)
- ácido fólico (vitamina B9)
- cobalamina (vitamina B12)
- ácido ascórbico (vitamina C)
- biotina (vitamina B8)
- protosoárina (vitamina B3)
Lipossolúveis
- Vitamina A
- Vitamina D
- Vitamina E
- Vitamina K
Tabela de Vitaminas
editar| Vitaminas Fontes Doenças provocadas pela carência (avitaminoses) | Funções no organismo | |
|---|---|---|
| a | Espinafre, leite, manteiga, cenoura, mamão e tomate. | problemas de visão, secura da pele, diminuição de glóbulos vermelhos. |
| d | Leite, atum, manteiga, óleo de fígado de peixe. | raquitismo e osteoporose
regulação do cálcio do sangue e dos ossos |
| K | Exemplo | deficiência na coagulação do sangue, hemorragias. atua na coagulação do sangue, previne osteoporose. |
| B1 | Pães, feijão, soja, ovos, fígado. | beribéri atua no metabolismo energético dos açúcares |
| B2 | Leite, queijo, iogurte, vegetais verdes folhosos, frutas, pão, cereais e vísceras | inflamações na língua, anemias, seborreia atua no metabolismo de enzimas, proteção no sistema nervoso. |
| 'B6 | Carnes, grãos integrais e levedo. | seborreia, anemia, distúrbios de crescimento crescimento, proteção celular, metabolismo de gorduras e proteínas, produção de hormônios |
| B12 | fígado, carnes anemia perniciosa | formação de hemácias e multiplicação celular |
| C | laranja, limão, abacaxi, kiwi, acerola, morango. | escorbuto atua no fortalecimento de sistema imunológico, aumenta a absorção do ferro pelo intestino. |
Ácidos nucleicos
editarOs ácidos nucléicos são macromoléculas compostas por monômeros denominados nucleotídeos descoberto por Johan Friedrich Miescher (foto) em 1869. De um ponto de vista bioquímico, os ácidos nucléicos têm a importante função de armazenar a informação gênica de uma célula e também é capaz de formar estruturas intracelulares. O ácido desoxirribonucléico (ADN) e ácido ribonucléico (ARN) são os principais ácidos nucléicos encontrados na terra.

Os ácidos nucléicos são normalmente encontrados na forma de fita simples ou dupla, mas estruturas com três ou mais fitas também são possíveis. Dentro da célula o DNA normalmente se encontra com dupla fita, no entanto, alguns vírus contem DNA de fita simples, o RNA encontra-se predominante na forma de fita simples o que permite que este dobre sobre si mesmo para formar estruturas terciárias de modo semelhante às proteínas. A diferença entre o DNA e RNA está em seus monômeros constituintes que será abordado em maiores detalhes mais adiante.

Índice
editar
|
Esta página é um esboço de química. Ampliando-a você ajudará a melhorar o Wikilivros. |
São as unidades fundamentais dos ácidos nucléicos. Ligam-se uns aos outros através de ligações fosfodiéster, formando cadeias muito longas com milhões de resíduos de comprimento. Além de participarem da estrutura dos ácidos nucléicos, os nucleotídeos atuam também como componentes na estrutura de coenzimas importantes no metabolismo oxidativo da célula, e como forma de energia química - ATP, por exemplo. Atuam ainda como ativadores e inibidores importantes em várias vias do metabolismo intermediário da célula.
By: Philipe do Vale Oliveira
Ácido Desoxirribonucleico
O ADN, sigla em português ou DNA, sigla em Inglês, é a molécula que contém as informações genéticas. É formado por quatro tipos de nucleotídeos e quatro tipos de bases nitrogenadas (adenina, timina, guanina e citosina) que irão formar moléculas de DNA distintas conforme a sequência e a quantidade desses nucleotídeos. No DNA contém informações gênicas que coordenam o desenvolvimento e funcionamento dos seres vivos e alguns vírus, as características hereditárias são passadas por meio dessa molécula, que tem o principal papel de armazenar as informações genéticas da célula.
Para o DNA possuir o formato de dupla hélice, os nucleotídeos formam pares de bases nitrogenadas e se unem através de pontes de hidrogênio, atrações frágeis que se formam apenas quando um hidrogênio está ligado a um átomo eletronegativo e se aproxima de outro átomo negativo, porém existem regras para essa formação em pares, a adenina só poderá se parear com a timina e vice-versa, já a guanina se pairará com a citosina e vice-versa. Portanto a quantidade de adenina no DNA é a mesma da timina e a quantidade de guanina será a mesma da citosina, sendo esta lógica denominada de relação Chargaff. As bases nitrogenadas possuem classificação em bases púricas (adenina e guanina) e bases pirimídicas (timina e citosina).
A duplicação do DNA é necessária devido à divisão celular, sendo denominada duplicação semiconservativa, pois ao ocorrer a separação das fitas de DNA pela enzima helicase, cada uma das fitas irá servir de molde para a construção de uma nova fita de DNA, o que faz a nova fita conservar uma parte do DNA antigo, portanto as duas novas moléculas de DNA terá em sua conformação uma parte do DNA antigo.
Os átomos que compõem as moléculas dos seres vivos vem dos carboidratos, lipídeos, proteínas e outras moléculas alimentares. Portanto, através dos alimentos se obtém matéria e energia do meio. METABOLISMO - Conjunto de reações químicas catalisadas por enzimas, que ocorrem no organismo. CATABOLISMO- é a conversão dos alimentos em moléculas mais simples e energia. ANABOLISMO- é a produção de grandes moléculas necessárias para as células a partir de pequenas moléculas e da energia liberada pelo catabolismo. O anabolismo também é conhecido como Biossíntese, porque sua sequência de reações sintetiza moléculas biologicamente importantes.
Faz parte da quarta etapa da respiração celular, denominada cadeia transportadora de elétrons e fosforilação oxidativa. Nesta etapa ocorrem fenômenos de oxidação-redução, oncd é libertada energia proveniente da cadeia transportadora de elétrons. Essa energia vai servir para fosforilar o ADP, formando ATP.
| Este livro ou módulo precisa ser formatado segundo o modelo wiki e/ou organizado conforme as convenções do Wikilivros. (discuta) Por favor ajude a formatar este módulo de acordo com as diretrizes estabelecidas no livro de estilo. Editor: considere colocar o mês e o ano da marcação. |
A informação genética é estocada no DNA por meio de um código (o código genético) no qual a seqüência de bases adjacentes determina a seqüência de aminoácidos no polipeptídeo codificado. O código genético, então é um dicionário que fornece a correspondência entre uma seqüência de bases nucleotídicas e uma seqüência de aminoácidos. Código genético=Receita genética (“livro de receitas de proteínas”). Em teoria, são possíveis variações quase infinitas na disposição das bases ao longo de uma cadeia polinucleotídica. Uma vez que existem 20 aminoácidos diferentes e apenas quatro bases diferentes de RNA, uma única base não pode especificar cada aminoácido. Em qualquer posição existem quatro possibilidades (A, T, C, G). Assim, existem 4 n combinações possíveis em uma seqüência de n bases. Dessa forma, aminoácidos específicos não podem ser determinados por duplas de bases, porque são possíveis apenas 16 ou 42
pares diferentes. Entretanto, se trincas de bases forem traduzidas em aminoácidos,
há 4 3 combinações possíveis de trincas, ou seja, 64 combinações podem ser obtidas, mais do que o suficiente para especificar cada aminoácido. O código genético foi decifrado em 1953. Watson e Crick demonstraram, de modo elegante e apurado, que o código consiste em códons, cada um composto por uma trinca de bases nitrogenadas (tripletes). Dos 64 códons (RNAm) possíveis, três indicam o fim de um gene, e são conhecidos como códons finalizadores (ou sem sentido) porque designam o termino da tradução do mRNA neste ponto. São o UAA, o UGA e o UAG. Os outros 61 especificam aminoácidos. Como existem apenas 20 aminoácidos essenciais, isto significa que a maioria dos aminoácidos pode ser especificada por mais de um códon. Por exemplo, a leucina e a arginina são especificadas por seis códons. Apenas a metionina e o triptofano são cada um deles especificado por um único códon O código genético é, portanto, dito “redundante” (ou degenerado). Embora um determinado aminoácido possa ser especificado por mais de um códon, cada códon só pode designar um aminoácido. Essa redundante descoberta é fundamental para, entre outras coisas, compreendermos que nem toda alteração no código genético leva a uma doença. Uma alteração de TTT para TTC, por exemplo, não deverá causar absolutamente nenhuma alteração no fenótipo de um individuo, porque ambos codificam o mesmo aminoácido. Porém há alterações na seqüência de ácidos nucléicos que podem resultar em um aminoácido inapropriado sendo inserido na cadeia polipeptídica, potencialmente causando uma doença ou mesmo a morte do organismo. Uma característica significativa do código genético é ser “universal”, ou seja, virtualmente todos os organismos vivos usam os mesmos códigos de DNA para especificar aminoácidos. Uma exceção conhecida a esta regra é a das mitocôndrias, as quais têm suas próprias moléculas de DNA extranuclear. Vários códons do DNA mitocondrial codificam aminoácidos diferentes dos códons do DNA nuclear. O código genético é extremamente conservado. Os mesmos trípletes correspondem aos mesmos aminoácidos, seja em seres humanos, seja em bactérias. A correspondência entre códons específicos e aminoácidos é conhecida como código genético. . Em síntese pode-se dizer que o Código Genético tem as seguintes características: - Especificidade - Universalidade - Redundância A informação genética está contida no DNA dos cromossomos dentro do núcleo celular, mas a síntese de proteínas, durante a qual a informação codificada no DNA é usada, ocorre no citoplasma. Devido à compartimentalização das células eucarióticas, a transferência de informação do núcleo para o citoplasma é um processo muito complexo. Enquanto o DNA se forma e se replica no núcleo da célula, ocorre no citoplasma a síntese de proteínas. A informação contida no DNA deve, portanto, ser transportada para o citoplasma, e assim usada para ditar a composição das proteínas. Isto envolve dois processos, transcrição e tradução. Resumidamente, o código do DNA é transcrito para o RNA mensageiro, que então deixa o núcleo para ser traduzido em proteínas. RNA Existem três tipos principais de RNA que participam do processo da síntese protéica: RNA ribossômico (RNAr), RNA de transferência (RNAt) e RNA mensageiro (RNAm). Assim como o DNA, esses três tipos de RNA são moléculas poliméricas não ramificadas, compostas de mononucleotídeos unidos por ligações fosfodiéster. Entretanto, eles diferem do DNA em vários aspectos; por exemplo, são consideravelmente menores, e contêm ribose em vez de desoxirribose e uracil em vez de timina. Os três principais tipos de RNA diferem um do outro em termos de tamanho, função e modificações estruturais especiais. RNA ribossômico: é encontrado em associação com uma série de proteínas diferentes, como componente dos ribossomos, as estruturas complexas que servem como sítios para a síntese de proteínas. No citosol eucariótico, existem quatro espécies de RNAr de tamanhos diferentes (28S, 18S, 5,8S e 5S). ["S" é a unidade Svedberg, relacionada ao peso molecular do composto]. Juntos constituem até 80% do RNA da célula. RNA de transferência: é a menor das três prinicipais moléculas de RNA (4S), tem entre 74 e 95 resíduos de nucleotídeos de tamanho, e forma de trevo. Existe no mínimo um tipo específico de molécula de RNAt para cada um dos 20 aminoácidos comumente encontrados nas proteínas. Juntos, eles constituem cerca de 15% do RNA da célula. As moléculas de RNAt contém bases incomuns que possuem extenso pareamento de bases intracadeia. Cada RNAt serve como um "adaptador", que transporta seu aminoácido específico ao sítio de síntese de proteínas. Lá, ele reconhece o termo do código genético que especifica a adição de seu aminoácido à cadeia peptídica em formação. RNA mensageiro: compreende somente cerca de 5% do RNA da célula e é o tipo mais heterogêneo de RNA em termos de tamanho. O RNAm transporta a informação genética do DNA ao citosol, onde é usado como molde para a síntese de proteínas. As características estruturais especiais do RNAm eucariótico incluem uma longa seqüência de nucleotídeos adenina (uma "cauda poli-A") no 3'-terminal da cadeia de RNA, mais uma "cabeça" no 5'- terminal ( cap 5’). TRADUÇÃO Um grande número de componentes são requeridos para a síntese da cadeia polipeptídica. Estes incluem todos os aminoácidos que são encontrados no produto final, o RNAm a ser traduzido, os RNAt, ribossomos funcionais, fontes de energia e fatores proteicos necessários à iniciação, elongação e terminação da cadeia polipeptídica. A tradução é o processo pelo qual o mRNA fornece um molde para a síntese de um polipeptideo. O mRNA é transportado do núcleo para o citoplasma, onde a seqüência de RNA é codificada, ou traduzida, para determinar a seqüência de aminoácidos na proteína que está sendo sintetizada. O mRNA não pode, entretanto, se ligar diretamente a aminoácidos. O RNA transportador (tRNA) fornece a ligação molecular entre a seqüência de bases do mRNA e a seqüência de bases da proteína. A ligação do anticódon do RNAt ao códon do RNAm segue as regras da ligação complementar e antiparalela - isto é, o códon do RNAm é lido de 5' para 3' por um acnódon p ti areado em orientação invertida (3'-5'). No citoplasma, o mRNA é traduzido em proteína pela ação de uma variedade de moléculas de tRNA, cada uma especifica para um determinado aminoácido. O RNAt tem a função de transferir os aminoácidos corretos para suas posições ao longo do molde de mRNA, para que sejam adicionados à cadeia polipeptídica crescente. O tRNA tem um sitio em sua ponta 3’ para a ligação de um aminoácido por uma ligação covalente. Na ponta oposta do trevo há uma seqüência de três nucleotídeos chamada de anticódon. Esta seqüência faz um pareamento complementar de bases com um códon apropriado no mRNA. O mRNA, portanto, especifica a seqüência de aminoácidos agindo por meio do tRNA. O local citoplasmático da síntese de proteínas é o ribossomo, que consiste em proteínas enzimáticas e RNA ribossomal (mais abundante). A função do rRNA é auxiliar a ligação do mRNA e do tRNA ao ribossomo. O ribossomo primeiramente liga-se a um sítio de iniciação na seqüência do mRNA. Este sítio consiste de um códon especifico, AUG, que especifica o aminoácido metionina. O ribossomo então liga o tRNA a sua superfície, de modo que possa haver pareamento entre o tRNA e o mRNA. O ribossomo move-se ao longo da seqüência de mRNA, códon por códon, no sentido usual 5’ para 3’. O ribossomo então desliza ao longo do mRNA a cada três bases, alinhando o códon seguinte para o reconhecimento por outro tRNA com o próximo aminoácido. À medida que cada códon é processado, um aminoácido é traduzido pela interação entre mRNA e tRNA. A ligação entre o códon e o anticódon coloca o aminoácido apropriado na posição seguinte no ribossomo para formação de uma ligação peptídica com a ponta carboxila e a cadeia poliptídica crescente. Neste processo, o ribossomo fornece uma enzima que catalisa a formação de ligações peptídicas covalentes entre aminoácidos adjacentes, resultando em um polipeptideo crescente. Quando o ribossomo chega a um códon finalizador na seqüência de mRNA, terminam a tradução e a formação de polipeptídeo. O terminal amino (NH2) do polipeptídeo correspondente à ponta 5’ do filamento de mRNA, e o terminal carboxila (COOH) corresponde à ponta 3’. Quando a síntese esta completa, o mRNA, o ribossomo e o polipetídeo se separam um do outro. O polipeptídeo é então liberado para o citoplasma. A tradução de um mRNA processado é sempre iniciada em um códon AUG, que especifica metionina. A metionina é, portanto, o primeiro aminoácido codificado (aminoterminal) de cada cadeia polipeptídica, embora em geral seja removido antes que a síntese da proteína esteja completa. O códon para metionina (iniciador AUG) estabelece a matriz de leitura do mRNA. Todo esse especializado e refinado processo pode ser dividido em três etapas básicas: Iniciação: A iniciação da síntese de proteínas envolve a reunião de componentes do sistema de tradução antes que ocorra a formação da ligação peptídica. Estes componentes incluem as duas subunidades ribossômicas, o RNAm a ser traduzido, o aminoacil-RNAt para metionina, especificado pelo códon iniciador AUG na mensagem, o GTP (o qual fornece energia ao processo) e fatores de iniciação que facilitam a montagem deste complexo de iniciação.
| Glossário |
|---|
|
A • B • C • D • E • F • G • H • I • J • K • L • M • N • O • P • Q • R • S • T • U • V • W • X • Y • Z |
Glossário
editarA
editar- Ácido forte – ácido que se dissocia totalmente na sua base conjugada e prótons quando em solução.
- Ácido fraco – ácido que se dissocia parcialmente na sua base conjugada e prótons quando em solução, estabelecendo-se um equilíbrio entre as espécies ionizadas e o ácido.
- Ácido gordo (Br. ácido graxo) - ácido composto por um grupo carboxilato ligado a uma cadeia de carbonos, geralmente longa e linear.
- Acilglicerol - composto de glicerol ligado a ácidos gordos através de ligações éster.
- Aeróbio – processo, mecanismo ou organismo que utiliza dioxigênio.
- Aeróbio facultativo – organismo que pode utilizar dioxigênio no seu metabolismo mas sobrevive sem este. Por exemplo: Escherichia coli.
- Aeróbio obrigatório – organismo que não sobrevive na ausência de dioxigênio, necessitando deste para o seu metabolismo energético. Por exemplo: humanos.
- Aldose - monossacarídeo contendo um grupo aldeído na sua estrutura.
- Aminoácido – molécula contendo um grupo amina e um grupo carboxílico; bloco básico de construção de péptidos e proteínas.
- Anabolismo – tipo de metabolismo em que é consumida energia para a síntese de biomoléculas necessárias à célula.
- Anaeróbio – processo, mecanismo ou organismo que não utiliza dioxigênio, podendo ou não ser destruído/inutilizado na presença deste.
- Anaeróbio aerotolerante – organismo que não utiliza dioxigênio no seu metabolismo mas é capaz de sobreviver na sua presença.
- Anfipático – diz-se do composto que possui uma parte hidrofílica e uma parte hidrofóbica. Por exemplo: fosfoacilgliceróis.
- Anfotérico – diz-se do composto que pode atuar como base e como ácido simultaneamente.
- Ångström – unidade de comprimento equivalente a 10−10m, muito usada na apresentação de distâncias entre átomos em estruturas tridimensionais de biomoléculas (especialmente proteínas).
- Anião – espécie química carregada negativamente (íon com carga elétrica negativa).
- Animalia – reino (divisão taxonômica) à qual pertencem todos os animais.
- Antibiótico – substância sintetizada por microrganismos (ou artificialmente) que destrói ou inibe o crescimento de outros microrganismos. Por exemplo: a penicilina.
- Anticodão – sequência de três nucleótidos de tRNA complementar a uma sequência de três nucleótidos de mRNA (codão) que identifica qual o aminoácido correspondente a esse codão.
- Antiparalelo – diz-se do arranjo das duas cadeias de DNA em dupla hélice por se entrelaçarem em sentidos opostos (ou seja, uma cadeia no sentido 5'–3' entrelaça-se com uma cadeia no sentido 3'–5').
- Apoenzima – parte proteica de uma enzima, ou seja, sem a presença de cofatores não proteicos.
- Átomo – a mais pequena partícula da matéria que conserva identidade química.
- Autotrofia – síntese de biomoléculas usando CO2 como fonte de carbono. Por exemplo, a fotossíntese é um processo autotrófico.
B
editar- Bactéria – microrganismo unicelular procariótico, que não possui organelas ou um núcleo definido.
- Beta-oxidação (β-oxidação) – via catabólica de oxidação de ácidos gordos, em que estes são degradados a pequenas moléculas orgânicas.
- Bicamada lipídica – camada de natureza dupla constituída por fosfoacilgliceróis, de tal modo que a parte hidrofílica ("cabeças") destes esteja em contato com o meio aquoso envolvente e a parte hidrofóbica ("caudas") esteja no interior da camada.
- Biorremediação – processo que emprega microrganismos com capacidade de transformar substâncias tóxicas em não-tóxicas. Usada, por exemplo, na limpeza de águas efluentes contaminadas.
- Biossíntese – síntese de moléculas usadas em processos bioquímicos. Requer o gasto de energia química, normalmente sob a forma de ATP.
C
editar- Cascata – em Bioquímica, refere-se à transmissão de informação através de várias reações, observando-se um aumento progressivo do sinal pretendido.
- Catabolismo – tipo de metabolismo em que existe degradação de biomoléculas mais ou menos complexas para haver libertação de energia.
- Catião – espécie química carregada positivamente (íon com carga elétrica positiva).
- Centro ativo – Zona de uma enzima onde o substrato se liga e é transformado a produto.
- Centro alostérico – Zona de uma enzima onde uma molécula (que não o substrato) se liga, modulando a atividade enzimática (estimulando-a, especificando-a ou inibindo-a).
- Cetose - monossacarídeo contendo um grupo cetona na sua estrutura.
- Cloroplasto – organela presente em células eucarióticas fotossintéticas, que contêm a maquinaria necessária para a fotossíntese.
- Constante de acidez – constante de dissociação de um ácido, definida como a razão entre o produto das concentrações de H+ e da base conjugada e a concentração do ácido. Dá uma medida da extensão da dissociação do ácido em solução.
D
editar- Desprotonação – perda de um ou mais prótons (H+) por uma molécula, dizendo-se que fica desprotonada.
E
editar- Energia de ativação – energia necessária para iniciar uma reação química. É diminuída na presença de enzimas.
- Enzima – proteína com características catalíticas, ou seja, que catalisa reações químicas em sistemas vivos.
- Epímero - monossacarídeo que difere de outro apenas na conformação quiral de um carbono.
- Equilíbrio químico – situação de estado estacionário de uma reação química reversível em que não existe uma variação líquida da concentração de reagentes ou de produtos, ou seja, em que a reação direta procede à mesma velocidade que a reação inversa.
F
editar- Fermentação alcoólica – processo metabólico em que o piruvato é reduzido pelo NADH a etanol.
- Fórmula de projeção de Fischer - forma de representação da estrutura de monossacarídeos em que o grupo carbonilo se desenha próximo do topo, ligações químicas desenhadas na vertical consideram-se atrás do plano e na horizontal acima do plano. É particularmente útil para determinar o tipo de enantiômero (D ou L) representado.
G
editar- Glícido – substância química composta por carbono, hidrogênio e oxigênio, que tem um papel de reserva/fonte de energia, estrutural ou de sinalização celular (quando ligado a proteínas membranares).
H
editar- Hexose - monossacarídeo contendo seis carbonos na sua estrutura.
- Heptose - monossacarídeo contendo sete carbonos na sua estrutura.
- Hidrofílico – diz-se do composto que possui afinidade para a água, sendo solúvel nesta. Por exemplo: glucose.
- Hidrofóbico – diz-se do composto que repele a água, sendo insolúvel nesta. Por exemplo: ácidos gordos.
- Holoenzima – enzima completa com cofatores não proteicos. O termo é aplicado quando é necessário distinguir esta da apoenzima.
I
editar- Íon – espécie química com carga elétrica, adquirida pelo ganho ânion ou perda cátion de elétrons.
- Isótopo – diz-se de átomos que, possuindo a mesma massa atômica, diferem no seu número atômico.
J
editarL
editar- Ligação de hidrogênio – ligação química fraca (23 kJ/mol) que se estabelece entre um átomo com alta eletronegatividade (como o oxigênio) e um átomo de hidrogênio.
- Ligação química – interação entre os elétrons de valência de átomos de modo a que estes atinjam uma configuração eletrônica de valência estável (de menor energia).
- Lipofílico – diz-se do composto que é solúvel num meio constituído por lípidos, como as membranas celulares (cf. hidrofóbico).
- Lípido ou lipídio - compostos orgânicos insolúveis em água com funções estruturais, de armazenamento de energia ou outras funções diversas, incluindo mensageiros intracelulares, alguns hormônios e pigmentos.
M
editar- Massa atômica – soma do número de prótons e de neutrões existentes num átomo.
- Membrana citoplasmática – bicamada lipídica que determina a divisão entre o interior da célula (meio intracelular) e o seu ambiente (meio extracelular), tendo entre outros um papel de compartimentação, regulação osmótica e transporte de substâncias.
- Molécula orgânica – designação geral dada a moléculas constituídas por pelo menos uma cadeia de átomos de carbono ligados entre si.
N
editar- Nomenclatura binomial – sistema de nomenclatura dos organismos vivos, criado por Lineu, para a designação de uma espécie por dois nomes, o primeiro designando o gênero a que pertence a espécie (em maiúscula) e o segundo sendo um epíteto específico (em minúscula). O nome deve ser destacado usando sublinhado ou itálico. Por exemplo: Helicobacter pylori (uma bactéria existente no estômago de parte da população humana).
- Número atômico – número de prótons de um átomo, que define a que elemento químico esse átomo pertence.
O
editarP
editar- Par conjugado ácido/base – par constituído por um ácido e respectiva forma desprotonada (base). Por exemplo: HCl/Cl-.
- Parede celular – invólucro exterior presente na maioria das células de plantas, fungos e procariontes que confere rigidez e forma à célula, protegendo-a também da entrada de substâncias nocivas e evitando a ruptura celular em condições hipotônicas.
- Pentose - monossacarídeo contendo cinco carbonos na sua estrutura.
- Peso atômico – o mesmo que (e mais corretamente designado como) massa atômica.
- pH – escala logarítmica que relaciona a concentração de iões H+ em solução aquosa com a acidez dessa solução.
- Ponte de hidrogênio – o mesmo que (e mais corretamente designada como) ligação de hidrogênio.
- Proteína transmembranar – diz-se da proteína que atravessa totalmente uma membrana, estabelecendo uma via de comunicação entre os dois compartimentos definidos por essa membrana. Têm geralmente uma função de sinalização celular ou transporte.
- Protonação – ganho de um ou mais prótons (H+) por uma molécula, dizendo-se que fica protonada.
Q
editar- Quimioautotrofia – tipo de autotrofia em que é usada a oxidação de compostos inorgânicos simples (por exemplo, nitritos) para a produção de energia química.
- Quimiosmose – processo de produção de ATP através de um gradiente de prótons, criado previamente através de uma membrana por transporte ativo.
- Quiralidade - diz-se das moléculas que, tendo a mesma composição química, diferem estruturalmente entre si como se fossem imagens de espelho.
- Quitina – homopolissacarídeo estrutural, constituído por N-acetilglucosamina. Encontra-se na parede celular de diversos fungos e é um componente essencial do exoesqueleto de artrópodes.
R
editar- Respiração aeróbia – denominação do processo global de catabolismo energético em que moléculas orgânicas são oxidadas através do ciclo dos ácidos tricarboxílicos e da fosforilação oxidativa a produtos como o CO2 e H2O.
S
editar- Solução aquosa – solução em que o solvente principal (ou o único solvente) é água. Todos os fluidos biológicos são soluções aquosas.
T
editar- Teoria celular – teoria desenvolvida por Matthias Schleiden e Theodor Schwann, em meados do século XIX, que afirma que todos os organismos são constituídos por células e estas são então a unidade fundamental de construção da vida.
- Tetrose - monossacarídeo contendo quatro carbonos na sua estrutura.
- Transporte ativo – tipo de transporte de espécies químicas através de membranas celulares contrariando o gradiente de concentração existente através dessa membrana. Requer energia (ATP) para ser efetuado e dá-se através de proteínas transmembranares.
- Triose - monossacarídeo contendo três carbonos na sua estrutura.
U
editarV
editar- Via anabólica – via metabólica cujo balanço global energético é negativo (é consumida energia) para a síntese de moléculas necessárias à célula.
- Via anfibólica – via metabólica que, em determinadas condições, serve para produzir energia e noutras condições para a síntese de metabolitos.
X
editarZ
editar
Referências
editarNeste módulo, são apresentadas as referências usadas na construção do livro de Bioquímica, sendo todas elas de leitura recomendada.
Gerais
editar- NELSON, David L., COX, Michael M., "Lehninger Principles of Biochemistry", 4ª edição, W. H. Freeman, 2005, ISBN 978-0716743392
- BLACK, Jacquelyn G., "Microbiology: principles and applications", 3ª edição, Prentice-Hall, New Jersey (E.U.A), 1996, ISBN 0-13-190745-X
- PURVES, William K., ORIANS, Gordon H., HELLER, H. Craig, "Life: The Science of Biology", 4ª edição, Sinauer Associates, Sunderland (E.U.A.), 1995, ISBN 0-7167-2629-7
- BROCK, William, H., "The Fontana History of Chemistry", Fontana Press, Londres (Reino Unido), 1992, ISBN 0-00-686173-3
Ligações externas
editar- Página oficial das normas de nomenclatura em Bioquímica da União Internacional de Bioquímica e Biologia Molecular (IUBMB).
- Página oficial das normas de nomenclatura em Química da União Internacional de Química Pura e Aplicada (IUPAC).